



Содержание
Перейти к:
https://doi.org/10.37489/2588-0519-2021-4-60-74
Перейти к:
Приём лекарственных средств (ЛС), потенциально, может приводить к повышению риска развития нежелательных реакций (НР), в том числе серьёзных, способствующих повышению риска смерти или развития состояний, потенциально повышающих смертность и/или заболеваемость, и/или становится причиной возникновения клинических проявлений, требующих обращения пациента за медицинской помощью или госпитализации. Такие осложнения фармакотерапии называют лекарственно-индуцированными заболеваниями (англ.: drug-induced diseases). Некоторые патологические состояния, такие как хроническая сердечная недостаточность (ХСН), являются потенциальными факторами риска лекарственно-индуцированных заболеваний (ЛИЗ) за счёт изменений фармакокинетики и фармакодинамики лекарственных средств. Например, после перорального приёма фозиноприла среднее значение Т1/2 у пациентов с ХСН II или III функционального класса составило 14,2 (±7,3) ч, а у здоровых лиц контрольной группы - 11,0 (±5,2) ч. Значения AUC per os и Cmax также были несколько выше у пациентов с сердечной недостаточностью (СН), чем у здоровых лиц, а Cl per os, наоборот, ниже. После внутривенного введения фозиноприла наблюдались сходные результаты. Другим примером является изменение абсорбции фуросемида у пациентов с декомпенсированной СН. Так, у пациентов с СН, по мере коррекции отёчного синдрома, наблюдаются снижение времени наступления максимальной концентрации препарата в сыворотке крови (Tmax ) на 27 % и увеличение Cmax на 29 %, что может свидетельствовать об уменьшении замедления скорости абсорбции (на 57 %). Поскольку фуросемид в основном выводится с мочой в неизменённом виде, наблюдаемые изменения Cmax и Tmax могли быть связаны с задержкой опорожнения желудка, снижением перистальтики кишечника или отёком стенки кишечника. Индивидуальный подбор дозы и режима дозирования с учётом особенностей фармакокинетики ЛС у пациентов с ХСН будет способствовать повышению качества жизни и профилактике потенциальных НР.
Переверзев А.П., Остроумова О.Д. Сердечная недостаточность как фактор риска развития нежелательных реакций. Часть 2: изменение фармакокинетики отдельных лекарственных средств. Качественная клиническая практика. 2021;(4):60-74. https://doi.org/10.37489/2588-0519-2021-4-60-74
Pereverzev A.P., Ostroumova O.D. Heart failure as a risk factor of adverse drug reactions. Part 2: potential changes in pharmacokinetics of some drugs. Kachestvennaya Klinicheskaya Praktika = Good Clinical Practice. 2021;(4):60-74. (In Russ.) https://doi.org/10.37489/2588-0519-2021-4-60-74
Приём лекарственных средств (ЛС), потенциально, может приводить к повышению риска развития нежелательных реакций (НР), в том числе серьёзных, способствующих повышению риска смерти или развития состояний, потенциально повышающих смертность и/или заболеваемость, и/или становится причиной возникновения клинических проявлений, требующих обращения пациента за медицинской помощью или госпитализации [1][2]. Такие осложнения фармакотерапии называют лекарственно-индуцированными заболеваниями (англ.: drug-induced diseases) [1][2]. Некоторые патологические состояния, такие как хроническая сердечная недостаточность (ХСН), являются потенциальными факторами риска лекарственно-индуцированных заболеваний (ЛИЗ) за счёт изменений фармакокинетики и фармакодинамики лекарственных средств [1][2].
Целью данной статьи является информирование специалистов практического здравоохранения в отношении отдельных лекарственных средств, приём которых у пациентов с ХСН может быть ассоциирован с изменениями фармакокинетики, требующими коррекции дозы или режима дозирования для профилактики потенциальных НР.
ИАПФ являются основной группой ЛС для лечения ХСН [3]. Продолжительность действия большинства иАПФ составляет около 24 ч, за исключением каптоприла, который действует около 6 ч [4]. Начало действия обычно быстрое (около 1 ч), большинство иАПФ (за исключением каптоприла и лизиноприла) являются пролекарствами, которые для оказания своего фармакологического действия должны метаболизироваться до активных метаболитов.
Каптоприл / Captopril. Каптоприл имеет достаточно большую биодоступность. Его Foral составляет 70–75 %, выводится каптоприл через печень и почки в одинаковой степени (по 50 %) [5]. Nishida M и cоавт. [6] изучали фармакокинетику каптоприла у 12 пациентов с СН (I–III ФК по NYHA) после приёма однократной дозы (12,5 мг) per os. Во всех 3 группах (согласно ФК CН) среднее значение Тmax составляло 2 часа, средние значения Cmax — 281 (±116), 261 (±151) и 274 (±122) нг/мл для классов I, II и III по NYHA соответственно. Средние значения Т1/2 — 2,79; 4,00 и 3,16 ч соответственно. Авторы отметили, что Тmax и Т1/2 каптоприла были более продолжительными по сравнению со значениями, указанными для здоровых добровольцев или пациентов с артериальной гипертензией (АГ). Авторы также выполнили дополнительный анализ, разделив пациентов на 2 группы согласно ФК СН по NYHA: группа I состояла из 7 пациентов с ФК I–II по NYHA, а группа II включала 5 пациентов с ФК III по NYHA. Было обнаружено, что Т1/2 каптоприла у пациентов I группы составило 2,79 ч, а у пациентов II группы — 4,00 ч. Авторы не сообщают о состоянии функции печени и почек у пациентов, вошедших в исследование [6].
Эналаприл / Enalapril. Эналаприл является пролекарством и метаболизируется до своего активного метаболита (эналаприлат) во время пресистемногометаболизма. Препарат отнесён к I классу BCS; абсорбция при приёме per os составляет не менее 61 % [7]. В своей работе Schwartz JB и соавт. [8] оценили концентрации эналаприла и эналаприлата в плазме крови у 8 пациентов с СН III–IV ФК по NYHA (группа наблюдения) и у 5 пациентов с лёгкой и умеренной АГ (группа сравнения) [8]. Пациенты с СН получали эналаприл с титрацией дозы — последовательно дозы 2,5; 5 и 10 мг один раз в сутки, пациентам с АГ были назначены более высокие дозы препарата (20 мг 2 раза в день или 40 мг один раз в день). По сравнению с пациентами с АГ у больных с СН Т1/2 было более продолжительным, как для эналаприла, так и для его метаболита. Значение Cl эналаприла при пероральном приёме у больных с СН также было ниже (0,6–0,7 л/мин.) по сравнению с пациентами с АГ (2,5–2,7 л/мин.). В обеих группах наблюдалось непропорциональное увеличение AUC per os эналаприла: с более чем трёхкратное при удвоении дозы препарата, чего не отмечено для эналаприлата (увеличение AUC было пропорционально увеличению дозы) [8].
Фозиноприл / Fosinopril. Фозиноприл отнесён к классу II по BCS, а его Foral составляет всего 30 % [49], что связано с неполным всасыванием, а не с эффектом первого прохождения [9]. Значения Foral фозиноприла у пациентов с СН существенно не отличаются от таковых у здоровых лиц [10]. Фозиноприл также является пролекарством и почти полностью метаболизируется до своего активного метаболита фозиноприлата. Он выводится в основном печенью, а также почками [9]. В открытом перекрёстном исследовании Kostis JB и соавт. [10] изучали ФК фозиноприла при внутривенном введении и при пероральном приёме [10] у 10 пациентов с СН II или III ФК по NYHA (ФВ ЛЖ ≤40 %) и у 10 здоровых лиц, сопоставимых по полу и возрасту (контрольная группа). Фозиноприл назначался в дозе 10 мг per os или 7,5 мг внутривенно. После перорального приёма фозиноприла среднее значение Т1/2 у пациентов с СН составило 14,2 (±7,3) ч, а у лиц контрольной группы — 11,0 (±5,2) ч. Значения AUC per os и Cmax также были несколько выше у пациентов с СН, чем у здоровых лиц, а Cl per os, наоборот, ниже. После внутривенного введения фозиноприла наблюдались сходные результаты [10].
Лизиноприл / Lisinopril. Лизиноприл отнесён к классу II по BCS. Его Foral составляет 25 %, при этом характерна большая межиндивидуальная вариабельность (от 6 до 60 %) [7][11]. Лизиноприл является активным веществом и не метаболизируется в печени; выводится с мочой в неизменённом виде. У пациентов со стабильной СН II–IV ФК по NYHA концентрация препарата снижается примерно на 16 % [12]. Gautam PC и соавт. [13] сравнили фармакокинетику ФК лизиноприла в 3 группах пациентов: молодые здоровые взрослые лица, пожилые здоровые взрослые лица и пациенты с СН [13]. Все участники принимали лизиноприл по 5 мг ежедневно в течение 7 дней, а образцы крови были взяты в дни 1 и 7. У пациентов с СН средние значения Cl лизиноприла при пероральном приёме были ниже по сравнению как с молодыми, так и с пожилыми здоровыми лицами [13].
Периндоприл / Perindopril. Периндоприл относится к классу I по BCS, Foral составляет 75–95 % [14]. Периндоприл является неактивным пролекарством и превращается в свой активный метаболит периндоприлат путём гидролиза в печени. Препарат характеризуется коротким Т1/2 (0,8–1 ч). Периндоприлат выводится преимущественно через почки, только 4–12 % пероральной дозы выводится с мочой в неизменённом виде, а почечный клиренс (Clrenal) составляет 170 мл/мин.; Т1/2 у здоровых людей — 3–10 ч [14]. Bellissant E и соавт. [15] изучали фармакокинетику периндоприла у 10 пациентов с СН III ФК по NYHA: как для периндоприла в неизменном виде, так и для его метаболита средние значения максимальной концентрации в крови (Cmax) и AUC у пациентов с СН были выше, чем у здоровых добровольцев. Также у пациентов с СН были выше значения Tmax и Т1/2. Среднее значение Cmax периндоприлата у пациентов с СН было в 3 раза выше, чем у здоровых добровольцев, а Т1/2 периндоприлата у пациентов с СН было значительно снижено — примерно до одной десятой от значений у здоровых субъектов, средняя AUC72 была сопоставимой. Эти данные позволяют предположить, что у пациентов с СН выведение периндоприлата может увеличиваться [15].
Хинаприл / Quinapril. Хинаприл относится к классу III по BCS, Foral составляет 50 % [16]. Хинаприл является пролекарством и после всасывания в кишечнике превращается в свой активный метаболит, хинаприлат. Squire IB и соавт. [17] изучали ФК хинаприла и его метаболита хинаприлата после однократного перорального приёма 2,5 мг у 12 пациентов с СН II или III ФК по NYHA. Среднее Тmax составило 2,6 (±1,2) ч для хинаприла и 3,6 (±0,8) ч для его метаболита. Соответствующие значения Cmax для хинаприла и хинаприлата составляли 49,7 (±30,9) и 51,0 (±22,8) нг/мл соответственно, AUC24 для хинаприлата — 422 (±259) нг•ч/мл. В данном исследовании не было контрольной группы здоровых лиц, поэтому сравнение проводилось косвенное, учитывались данные других исследований, в которых участвовали здоровые добровольцы: AUC72 хинаприлата у здоровых лиц cоставляла 288 нг•ч/мл, что означает, что у пациентов с СН AUC выше, чем у здоровых людей. Фармакокинетика хинаприла после многократного приёма изучалась ещё в одном исследовании [18]: пациенты с СН II или III ФК по NYHA (n=12) принимали хинаприл в дозе 10 мг 2 раза в сутки в течение 4 недель. Средняя Cmax хинаприлата составляла 362 нг/мл, Тmax — 1,88 часа, AUC12 — 1706 мкг•ч/мл, Т1/2 — 3,7 ч. Была выявлена статистически значимая взаимосвязь между ФВ ЛЖ и Т1/2 (r2 = 0,57, p = 0,005).
Рамиприл / Ramipril. Рамиприл относится к I классу по BCS, Foral составляет 50–60 % [14][19]. Рамиприл является пролекарством и почти полностью превращается в печени в свой активный метаболит рамиприлат [14]. Рамиприлат выводится в основном через почки, Cl — 40,3 мл/мин. [14], а также с желчью в размере, равном 1/3 от количества, выделяемого с мочой в течение 24 часов после перорального приёма [14].
Kondo K и соавт. [65] в своей работе изучали ФК рамиприла в дозе 5 мг один раз в сутки в течение 15 дней у здоровых добровольцев: средняя Cmax и AUC в интервале между дозами на 15-й день для рамиприла и рамиприлата составили 18,8 против 15,5 нг/мл и 39,2 против 102 нг•ч/мл соответственно. Следовательно, косвенное сравнение позволяет говорить об увеличении значений Cmax и AUC рамиприлата у пациентов с СН [20].
В таблице 1 систематизирована информация о потенциальных изменениях фармакокинетики иАПФ у пациентов с ХСН, полученная из открытых литературных источников [6][8][10][13–15][17][18][21][22][26–28].
В лечении ХСН применяются 3 представителя данного класса (кандесартан, лозартан и валсартан) [5].
Кандесартан / Candesartan. Кандесартан цилексетил отнесён к IV классу BCS и имеет низкий и изменчивый показатель Foral — от 15 до 42 % [14]. Кандесартан цилексетил является пролекарством, быстро и полностью метаболизируется до кандесартана, активного компонента, во время пресистемного метаболизма в тканях ЖКТ и печени. Выводится в основном через почки.
Anpo Y и соавт. [21] изучали фармакокинетику кандесартана у 5 пациентов с СН II или III ФК по NYHA после перорального приёма в дозе 4 мг. Средние значения Cmax, AUC per os и Т1/2 составили 57 (±22) нг/мл, 825 (±514) нг•ч/мл и 12,0 (±2,9) ч соответственно [21]. Прямых сравнений фармакокинетических параметров кандесартана у пациентов с наличием и отсутствием СН не проводилось. Однако значения фармакокинетических параметров, полученные Anpo Y и соавт., в значительной степени сопоставимы с их значениями, полученными у пожилых пациентов с АГ [22]. Так, Cmax кандесартана составила 57 (±12) нг/мл, AUC per os48 — 577 (±132) нг•ч/мл и Т1/2 — 11,7 (±2,8) ч [22]. Следовательно, фармакокинетика кандесартана, по-видимому, не зависит от наличия нетяжёлой СН, если, конечно, у пациентов нет сопутствующего нарушения функции почек [21][22].
Лозартан / Losartan. Лозартан отнесён ко II классу BCS; он имеет довольно низкую Foral (25–35 %) [23]. Лозартан интенсивно метаболизируется до 5-карбоновой кислоты E3174, причём метаболит выводится почечным и непочечным путями в равной степени.
Lо MW и соавт. [24] исследовали фармакокинетику лозартана у 11 пациентов с CН и ФВ ≤45 % в ходе открытого перекрёстного исследования. Пациенты получали лозартан в дозе 10 мг внутривенно и в дозе 50 мг перорально в течение 7—8 дней. После повторного перорального приёма лозартана средняя AUC per os составила 577,2 нг•ч /мл (±267,1), Т1/2 — 3,3 (±1,4) ч, Cmax — 223,3 (±199,4) нг/мл, Тmax — 1,31 (±0,93) ч, Foral — 35,5 % (95 % доверительный интервал (ДИ) 29,3–43,0), Clrenal — 42,6 (±25,5) мл/мин. Для метаболита E3174 Cmax была аналогичной (222,9±91,6 нг/мл), Тmax и Т1/2 увеличивались на 4,5 (±1,1) и 7,6 (±1,5) ч соответственно, с более высокой AUC per os (2262,8±1,225,4 нг•ч/мл), а Clrenal был ниже (18,1±5,9 мл/мин.). Авторы сравнили полученные ими данные с результатами ранее опубликованного исследования фармакокинетики лозартана у здоровых добровольцев: cредние значения Clrenal для лозартана и E3174 у пациентов с СН были ниже, чем у здоровых взрослых лиц (72 и 25,9 мл/мин. соответственно), также у пациентов с СН оказались ниже и значения AUC по сравнению со здоровыми добровольцами (476 и 1 915 нг•ч/мл соответственно для лозартана и E3174) [24].
Валсартан / Valsartan. Валсартан отнесён к IV классу BCS и имеет низкий показатель Foral (25 %). Он не имеет активных метаболитов и выводится в основном (89 %) с желчью в неизменённом виде [25]. Поскольку клиренс (2,2 л/ч (37 мл/мин)) в значительной степени обусловлен выведением печенью, низкий показатель Foral, скорее всего, связан с неполной абсорбцией препарата в кишечнике.
Фармакокинетика валсартана была исследована Prasad PP и соавт. [26] в открытом исследовании с участием 20 пациентов с СН II или III ФК по NYHA (ФВ ЛЖ ≤40 %) [71]. Пациенты получали валсартан в дозах 40, 80, а затем 160 мг 2 раза в сутки (каждый вариант дозы в течение 7 дней). Фармакокинетические параметры оценивали в последний день приёма каждой дозы (то есть 7, 15 и 21-й дни). Значения Cmax, Тmax, минимальной концентрации (Cmin), AUC per os и Т1/2 увеличивались пропорционально дозе (80 мг в 2 раза больше, чем 40 мг; 160 мг в 2 раза больше, чем 80 мг). Для дозы 40 мг средние или медианные значения Cmax, Тmax (медиана), Cmin, AUC per os и Т1/2 составляли 1,94 (±1,0) мкг/мл, 3 ч, 0,47 (±0,3) мкг/мл, 13,12 (±7,2) мкг•ч/мл и 5,2 (±1,9) ч соответственно. Для валсартана 80 мг соответствующие значения составляли 3,95 (±2,3) мкг/мл, 2,5 ч, 1,05 (±0,8) мкг/мл, 25,94 (±15,7) мкг•ч/мл и 6,5 (±2,4) ч. Для валсартана в дозе 160 мг значения указанных фармакокинетических параметров составили: 6,40 (±3,2) мкг/мл, 3 ч, 1,98 (±1,6) мкг/мл, 43,54 (±25,9) мкг•ч/мл и 6,6 (±3,9) ч соответственно. Возраст больных, ФК СН по NYHA и масса тела не оказали значимого влияния на фармакокинетику валсартана [26].
Сопоставимые фармакокинетические данные были получены у 6 здоровых добровольцев японцев, которые принимали 160 мг валсартана перорально один раз в день в течение 7 дней [27]. Средние значения для Cmax, Тmax, AUC per os24 и Т1/2, полученные на 7-й день, составили 3,72 (±0,63) мкг/мл, 3 ч (медиана), 21,6 (±6,9) мкг•ч/мл и 5,0 (±0,9) ч соответственно. По-видимому, значения фармакокинетических параметров валсартана (80 мг 2 раза в cутки) у пациентов с СН аналогичны таковым у здоровых добровольцев [27].
В таблице 1 систематизирована информация о потенциальных изменениях фармакокинетики БРА у пациентов с ХСН, полученная из открытых литературных источников [6][8][10][13][15][17][18][21][25–28].
Таблица 1
Исследования по изучению фармакокинетики блокаторов ренин-ангиотензин-альдостероновой системы у пациентов с СН [6][8][10][13][15][17][18][21][25–28]
Table 1
Summary of pharmacokinetic studies of renin-angiotensin-aldosterone blockers in patients with heart failure [6][8][10][13][15][17][18][21][25–28]
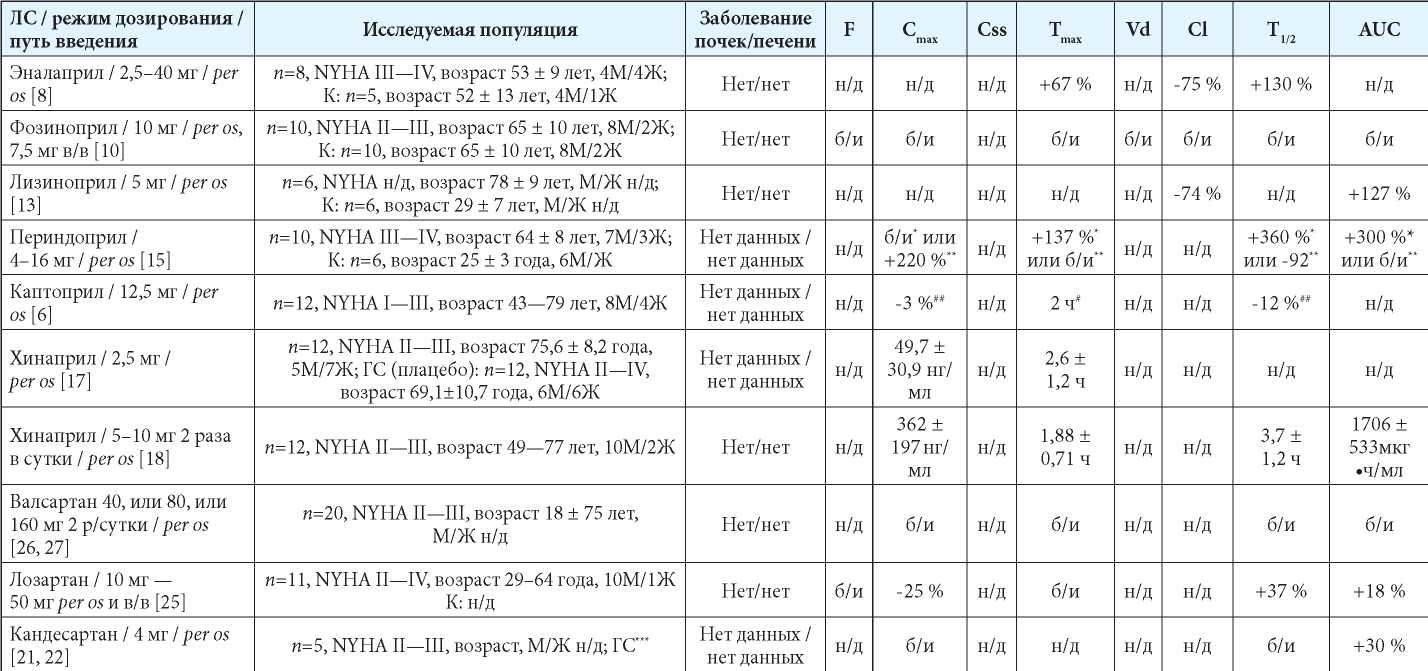
Примечания: * — периндоприл; ** — периндоприлат; *** — в группу сравнения вошли пациенты пожилого и старческого возраста с артериальной гипертензией без ХСН. Прямых сравнительных исследований кандесартана со здоровыми добровольцами не проводилось; # — среднее значение у пациентов всех трёх ФК по NYHA; ## — снижение в группе пациентов с IV ФК по NYHA относительно II ФК по NYHA; б/и — без статистических значимых изменений; в/в — внутривенно; ГС — группа сравнения; Ж — женщины; К — контроль (группа контроля); М — мужчины; н/д — нет данных; AUC — area under curve (площадь под кривой); Cl — клиренс; Cmax — максимальная концентрация в сыворотке крови; Css — равновесная концентрация в сыворотке крови; F — биодоступность; NYHA — New York Heart Association Functional Classification (функциональная классификация сердечной недостаточности Нью-Йоркской ассоциации сердца); T1/2 — период полувыведения; Tmax — время достижения максимальной концентрации в сыворотке крови; Vd — объём распределения.
Notes: * — perindopril; ** — perindoprilat; *** — the comparison group included elderly patients with arterial hypertension without CHF. Direct comparative studies of candesartan with healthy volunteers have not been conducted; # — mean value in patients of all three FCs according to NYHA; ## — decrease in the group of patients with FC IV according to NYHA relative to FC II according to NYHA; б/и — no statistically significant changes; в/в — intravenously; ГС — comparison group; Ж — women; K — control (control group); M — men; н/д — no data; AUC — area under curve; Cl — clearance; Cmax — maximum concentration in blood plasma; Css — steady-steate concentration; F — bioavailability; NYHA — New York Heart Association Functional Classification; T1/2 — half-life; Tmax — time to reach maximum concentration in blood plasma; Vd — the volume of distribution.
Для лечения СН рекомендованы 4 препарата данного класса: бисопролол, карведилол, метопролола сукцинат и небиволол [3].
Бисопролол / Bisoprolol. Бисопролол (селективный β1-адреноблокатор) у здоровых людей имеет высокую Foral — 84–92 % [29]. Препарат отнесён к III классу BCS. Клиренс составляет 14,2–15,6 л/ч
(230–260 мл/мин.), в которые почечный и печёночный Cl вносят примерно равный вклад [14]. Nikolic VN и соавт. [30] изучали изменения фармакокинетики бисопролола у 61 пациента с СН II–III ФК по NYHA и обнаружили снижение клиренса препарата при его пероральном приёме на 25–35 % (7,9 л/ч, или 131 мл/мин) по сравнению со здоровыми субъектами [30].
Карведилол / Carvedilol. Карведилол VN неселективный β-адреноблокатор с α-адреноблокирующим действием [14]. Это рацемическая смесь, в которой S (-) энантиомер обладает неселективной β1- и β2-блокирующей активностью, а R (+) энантиомер имеет равную α- и β-адреноблокирующую активность. Карведилол отнесён ко II классу BCS. Биодоступность карведилола низкая (около 25 %) из-за выраженного пресистемного метаболизма [14]. Карведилол подвергается интенсивному метаболизму в печени через ферменты цитохрома P450, в первую очередь CYP2D6 и CYP2C9; при пероральном приёме его Cl снижается у пациентов с циррозом печени и пациентов с генетическим полиморфизмом CYP2D6 [14]. Сообщалось также об увеличении AUC pеr os для карведилола у пациентов с почечной дисфункцией [14].
Tenero D и соавт. [31] исследовали фармакокинетику карведилола у 22 пациентов мужского пола с CН III или IV ФК по NYHA в открытом несравнительном исследовании [31]. Карведилол назначался с титрацией дозы (6,25–50 мг) 2 раза в день, шаг титрации — 7 дней. Выявлено, что значения как AUC per os, так и Cmax для одной и той же дозы у пациентов с СН IV ФК были выше, чем у пациентов с III ФК СН: для дозировок 6,25; 12,5; 25 и 50 мг средние значения Cmax у пациентов с СН III ФК составили 22,1 (±8,7), 40,9 (±18,5), 96,2 (±43,5) и 198 (±84) нг/мл соответственно; cоответствующие значения у пациентов с СН IV ФК составляли 30,9 (±33,9), 63,9 (±39,3), 119 (±88) и 212 (±143) нг/мл соответственно. Аналогичные результаты были получены для AUC per os, при этом более высокие значения AUC per os были зарегистрированы у пациентов с СН IV ФК (как минимум на 50 % выше), чем у пациентов с СН III ФК. Аналогичная картина наблюдалась для каждого из энантиомеров, при этом пациенты с СН IV ФК имели более высокие значения как Cmax, так и AUC per os. Наибольшее увеличение наблюдалось для AUC per os для R (-) энантиомера (≥50 %). Не было обнаружено значимых различий между Тmax, за исключением доз 6,25 и 12,5 мг [31].
Nikolic VN и соавт. [32] изучали ФК карведилола у 52 пациентов европеоидной расы с СН II ФК по NYHA (79 %) и III ФК по NYHA (21 %), получавших карведилол перорально в течение длительного времени. Авторы выявили, что Cl препарата составил, в среднем, 43,8 л/ч и был статистически значимо ассоциирован с такими факторами, как масса тела, совместное введение дигоксина и курение. Cl R (+) и S (-) энантиомеров карведилола также был значительно ниже по сравнению со здоровыми лицами [32]. Horiuchi I и соавт. [33] определяли пиковые и минимальные уровни R (+) и S (-) энантиомеров карведилола в образцах крови, взятых у 24 японских пациентов с СН, длительно лечившихся карведилолом. Дозы карведилола варьировали от 1,25 до 20 мг в сутки, при этом большинство пациентов (n=22) получали суточную дозу карведилола однократно. Cmax R (+) энантиомера в плазме крови пациентов была выше, чем S (-) энантиомера. Взаимосвязи Cl обоих энантиомеров карведилола при пероральном приёме с возрастом пациентов, этиологией СН и наличием аллеля CYP2D6*10 не обнаружено. Авторы сравнили результаты этого исследования с данными, полученными на здоровых добровольцах, и пришли к выводу, что Cl обоих энантиомеров карведилола при пероральном приёме составляет 25–29 % от значений, полученных у здоровых добровольцев с наличием аллеля CYP2D6. Это свидетельствует о том, что активность метаболизма карведилола при СН снижается [33].
В отличие от этих результатов, Saito M и соавт. [34] сообщили о снижении Cl карведилола при пероральном приёме у японских пациентов с СН (n=40) с наличием определённых генотипов CYP2D6 [34]. Из 56 пациентов, включённых в их исследование, генотипирование CYP2D6 было выполнено у 40 пациентов. У пациентов с аллелями CYP2D6 *1/*5, *5/*10 или *10/*10 Cl обоих энантиомеров карведилола при пероральном приёме был ниже, чем у пациентов с аллелями *1/*1 или *1/*10. Средние значения Cl для R (+) энантиомера составляли 0,23; 0,33 и 0,42 л/ч/кг соответственно в первой группе по сравнению с 0,59 и 0,64 л/ч/кг соответственно во второй группе. Соответствующие значения для S (-) энантиомеров составляли 0,40; 0,51 и 0,90 л/ч/кг (первая группа) соответственно, по сравнению с 1,07 и 1,12 (0,45) л/ч/кг (вторая группа) соответственно. На Cl карведилола при пероральном приёме оказывали влияние масса тела пациента и уровень кислого α1-гликопротеина, увеличивая его и уменьшая соответственно [34].
Метопролол / Metoprolol. Метопролол отнесён к классу I по BCS. Его Foral низкая (50 %) из-за выраженного эффекта первого прохождения. Метопролол выводится в основном за счёт метаболизма в печени, и менее 5 % введённой внутривенно дозы выводится с мочой в неизменённом виде [14].
Taguchi М и соавт. [35] изучали влияние генетического полиморфизма CYP2D6 на Cl метопролола при пероральном приёме у 34 японских пациентов, у 5 из которых была СН (4 пациента с II ФК по NYHA и один пациент с III ФК), и обнаружили, что наличие аллеля CYP2D6 *10 и возраст (>70 лет) были статистически значимо взаимосвязаны с Cl, а наличие СН — нет [35].
Laer S и соавт. [36] не выявили статистически значимых различий в фармакокинетике карведилола и метопролола между пациентами с наличием и отсутствием СН (в отсутствие нарушений функции почек и печени), за исключением значительно более короткого (на 44 %) Т1/2 у больных с СН.
Небиволол / Nebivolol. Небиволол быстро всасывается после приёма внутрь, но характеризуется крайне вариабельной биодоступностью (12–96 %) при пероральном приёме из-за выраженного пресистемного метаболизма [14]. Препарат метаболизируется преимущественно CYP2D6 до нескольких активных метаболитов, CYP2D6 подвержен генетическому полиморфизму, что приводит к вариабельному метаболизму небиволола [37]. Биодоступность небиволола при пероральном приёме может составлять от 12 % у пациентов с «быстрым» метаболизмом и до 96 % у пациентов с «медленным» метаболизмом. Из-за его интенсивного метаболизма в печени пациентам с умеренной печёночной недостаточностью следует начинать терапию небивололом в стартовой дозе 2,5 мг в день [14]. Метаболизм небиволола в печени является его основным путём выведения, при этом менее 0,5 % препарата в неизменённом виде выводится через почки. У пациентов с «быстрым» метаболизмом 38 % общей дозы небиволола выводится с мочой и 44 % — с калом, а с «медленным» метаболизмом — 66 % и 13 % с мочой и калом соответственно [14]. Т1/2 небиволола составляет примерно 10 часов у пациентов с «быстрым» метаболизмом и 32 ча са — с «медленным» метаболизмом. Cl небиволола значительно снижен у пациентов с тяжёлым нарушением функции почек, а при КК<20 мл/мин. препарат противопоказан [14].
Специальных исследований влияния СН на ФК небиволола на момент подготовки обзора не обнаружено, однако, учитывая тот факт, что СН может снижать активность CYP2D6 [36], можно предположить повышение риска передозировки и развития НР (брадикардия и др.) у пациентов, принимающих небиволол. Теоретически для профилактики данного осложнения фармакотерапии необходимо начинать терапию с минимальных доз небиволола, постепенно повышая до терапевтических, ориентируясь на частоту сердечных сокращений (ЧСС) [14].
В таблице 2 систематизирована информация о потенциальных изменениях фармакокинетики β-адреноблокаторов у пациентов с ХСН, полученная из открытых литературных источников [30–36].
Таблица 2
Исследования по изучению фармакокинетики β-блокаторов у пациентов с СН [30–36]
Table 2
Summary of studies of pharmacokinetics of β-blockers in patients with heart failure [30–36]
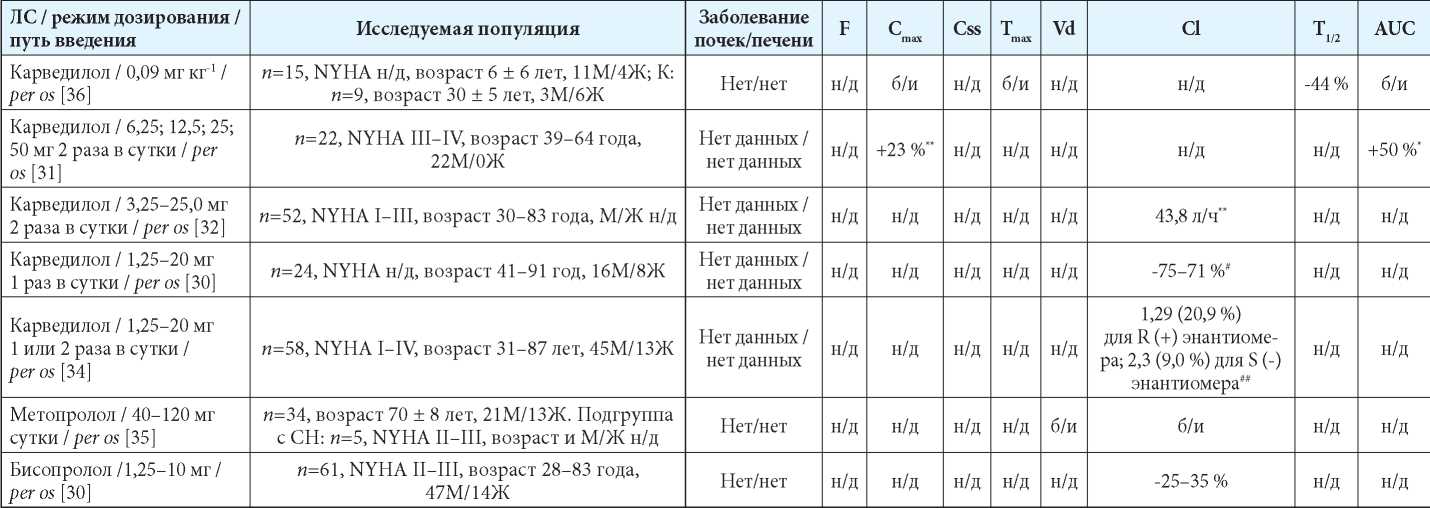
Примечания: Специальных исследований влияния СН на фармакокинетику небиволола в процессе подготовки статьи выявлено не было. Теоретически возможно повышение риска передозировки и развития нежелательных реакций вследствие снижения активности цитохрома CYP2D6, вызванной хронической сердечной недостаточностью. * — пациенты с NYHA IV относительно пациентов с NYHA III; ** — среднее значение; # — для R- и S-энантиомеров / ## — среднее значение Cl/F л/ч/кг (относительная стандартная ошибка, %); б/и — без статистически значимых изменений; Ж — женщины; К — контроль (группа контроля); М — мужчины; н/д — нет данных; СН — сердечная недостаточность; AUC — area under curve (площадь под кривой); Cl — клиренс; Cmax — максимальная концентрация в сыворотке крови; Css — равновесная концентрация в сыворотке крови; F — биодоступность; NYHA — New York Heart Association Functional Classification — функциональная классификация сердечной недостаточности Нью-Йоркской ассоциации сердца; T1/2 — период полувыведения; Tmax — время достижения максимальной концентрации в сыворотке крови; Vd — объём распределения.
Notes: No specific studies of the effect of HF on the pharmacokinetics of nebivolol were identified during the preparation of the article. It is theoretically possible to increase the risk of overdose and the development of adverse reactions due to a decrease in the activity of cytochrome CYP2D6 caused by chronic heart failure. * — patients with NYHA IV versus patients with NYHA III; ** — mean; # — for R (-) and S (-) enantiomers / ## — mean value of Cl / F l / h / kg (relative standard error, %); б/и — no statistically significant changes; Ж — women; K — control (control group); M — men; н/д — no data; AUC — area under curve; Cl — clearance; Cmax — maximum concentration in blood plasma; Css — steady steate concentration; F — bioavailability; NYHA — New York Heart Association Functional Classification; T1/2 — half-life; Tmax — time to reach maximum concentration in blood plasma; Vd — the volume of distribution.
В РКИ была продемонстрирована эффективность АМКР в снижении смертности и госпитализаций у пациентов с хронической СН с низкой ФВ [3]. Фармакокинетика эплеренона у пациентов с СН изучалась в единственном исследовании [38], а работ по исследованию фармакокинетики спиронолактона при СН на момент подготовки обзора в литературе не найдено.
Спиронолактон / Spironolactone. Foral спиронолактона составляет 80–90 %. В сыворотке крови 88 % препарата находится в связанном с белками (преимущественно альбумином) состоянии. Препарат активно метаболизируется в печени с образованием серосодержащих фармакологически активных метаболитов, T1/2 — 1,3–2 ч. Специальных исследований влияния СН на фармакокинетику спиронолактона в литературе не обнаружено. С учётом малого объёма распределения (0,05 л/кг), высокой биодоступности, возможно применять препарат даже у пациентов с тяжёлой почечной недостаточностью (до СКФ 10 мл/мин.), развитие клинически значимых изменений фармакокинетики маловероятно [14].
Эплеренон / Eplerenone. Эплеренон отнесён к классу II по BCS. Точное значение Foral не известно, но, по данным разных исследований [39], она колеблется в диапазоне 32–67 % [39]. Эплеренон интенсивно метаболизируется, и менее 5 % принятой дозы выводится с мочой в неизменённом виде. Активных метаболитов в настоящее время не обнаружено. Среднее значение Cl при пероральном приёме составляет 10 л/ч (167 мл/мин.), а средний Clrenal составляет 0,07–0,15 л/ч (1–2,5 мл/мин.) [40]. Фармакокинетика эплеренона изучалась у 8 пациентов с СН (II–IV ФК по NYHA), принимавших эплеренон в дозе 50 мг, и у 8 здоровых людей контрольной группы (группы были сопоставимы по полу, возрасту и массе тела), и было выявлено, что у пациентов с СН значения AUC per os и Cmax были выше на 38 и 30 % соответственно, чем у здоровых лиц [41].
В таблице 3 систематизирована информация о потенциальных изменениях фармакокинетики АМКР у пациентов с ХСН, полученная из открытых литературных источников [41–49].
Таблица 3
Исследования по изучению фармакокинетики диуретиков у пациентов с СН [41–49]
Table 3
Summary of Studies of pharmacokinetics of diuretics in patients with HF [41–49]
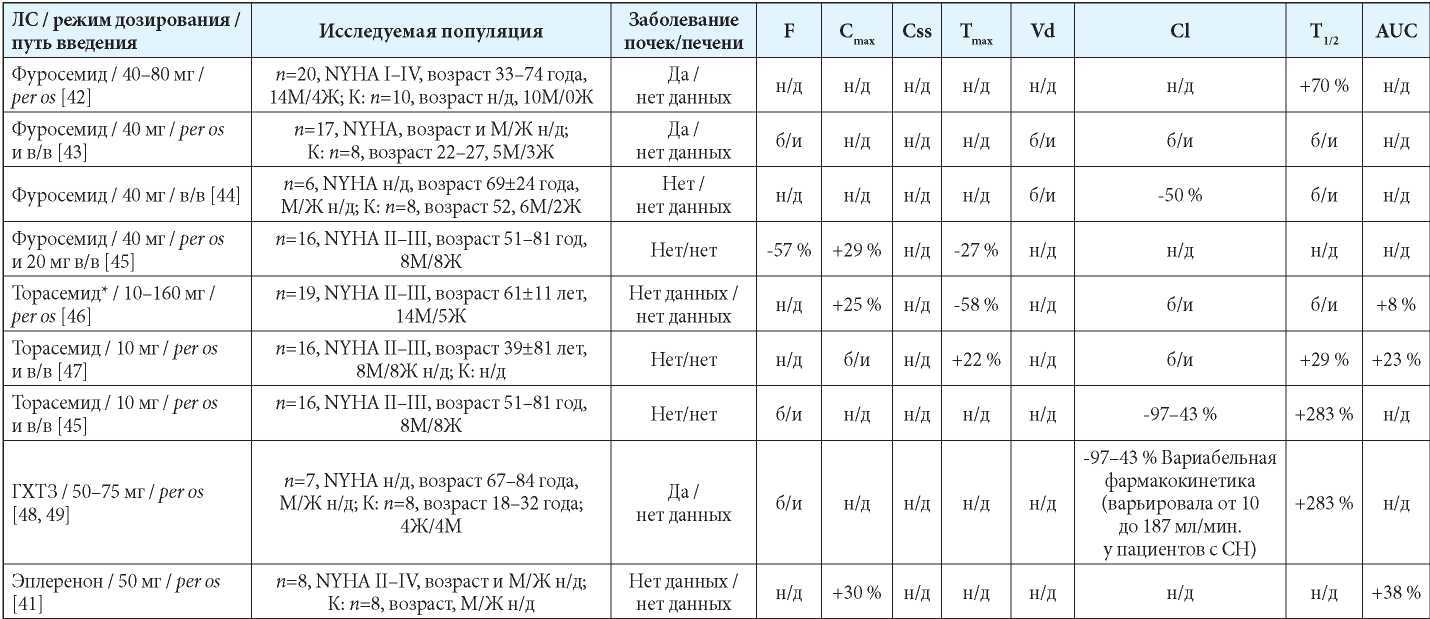
Примечания: Специальных исследований влияния СН на фармакокинетику спиронолактона в литературе на момент подготовки статьи не найдено. Теоретически клинически значимые изменения фармакокинетики у пациентов с СН маловероятны. б/и — без статистически значимых изменений; в/в — внутривенно; ГХТД — гидрохлортиазид; Ж — женщины; К — контроль (группа контроля); М — мужчины; н/д — нет данных; СН — сердечная недостаточность; AUC — area under curve (площадь под кривой); Cl — клиренс; Cmax — максимальная концентрация в сыворотке крови; Css — равновесная концентрация в сыворотке крови; F — биодоступность; NYHA — New York Heart Association Functional Classification (функциональная классификация сердечной недостаточности Нью-Йоркской ассоциации сердца); Tmax — время достижения максимальной концентрации в сыворотке крови; T1/2 — период полувыведения; Vd — объём распределения.
Notes: Special studies of the effect of HF on the pharmacokinetics of spironolactone have not been found in the literature at the time of preparation of the article. Th eoretically; clinically significant changes in pharmacokinetics in patients with heart failure are unlikely. б/и — no statistically significant changes; в/в — intravenously; ГХТД — hydrochlorothiazide; Ж — women; K — control (control group); M — men; н/д — no data; HF-heart failure; AUC — area under curve; Cl — clearance; Cmax — maximum concentration in blood plasma; Css — steady-steate concentration; F — bioavailability; NYHA — New York Heart Association Functional Classification; T1/2 — half-life; Tmax — time to reach maximum concentration in blood plasma; Vd — the volume of distribution.
Петлевые диуретики (фуросемид и торсемид) часто используются при лечении СН [3]. Механизм действия данных ЛС обусловлен связыванием с транспортёрами натрия-калия-хлора в петле Генле и ингибированием реабсорбции этих ионов [14]. Фуросемид отнесён к IV классу BCS и имеет низкую растворимость и мембранную проницаемость, а буметанид и торсемид — ко II и III классу BCS соответственно. Все они начинают действовать в течение 30–60 минут после введения, при этом Т1/2 составляют 60–90 минут, 2 часа и 3,5 часа для буметанида, фуросемида и торсемида соответственно. Фуросемид в основном выводится почками (фракция препарата, которая выводится почками в неизменном виде, — FR = 0,6–0,9), а его Cl и Vd у здоровых людей составляют 2 мл/мин./кг (140 мл/мин./70 кг) и 0,2 л/кг соответственно [14]. Поскольку его почечный Cl намного превышает СКФ, должна иметь место активная секреция в почечных канальцах. T1/2 фуросемида колеблется от 0,5 до 2 часов. Напротив, торсемид подвергается метаболизму в печени (FR=0,21) как в активные, так и в неактивные метаболиты [14]. Почечный Cl торсемида составляет от 6,4 до 13 мл/мин., его Т1/2 колеблется от 3 до 6 часов и увеличивается у пациентов с печёночной или почечной недостаточностью [14]. Фуросемид плохо абсорбируется в ЖКТ, а его Foral может варьировать от 12 до 112 % [50]. Напротив, Foral буметанида и торсемида близки к 100 % [14].
ФК фуросемида и торсемида у пациентов с СН оценивалась в небольшом перекрёстном исследовании: 16 пациентов с СН (II или III ФК по NYHA) были рандомизированы для однократного внутривенного введения либо фуросемида 20 мг, либо торсемида 10 мг [51]. Для фуросемида средние значения Cl, кажущийся объём распределения в равновесном состоянии (Vss) и Т1/2 составляли 138 мл/мин., 9,8 л и 1,5 ч соответственно. Для торасемида значения вышеуказанных параметров составили 36,1 мл/мин., 10,4 л и 6,3 ч соответственно. Эти фармакокинетические параметры в значительной степени сопоставимы с таковыми у здоровых лиц [51–53].
У пациентов с декомпенсированной СН абсорбция фуросемида будет замедленной и неустойчивой по сравнению со здоровыми людьми [45]. Vasko MR и соавт. [45] продемонстрировали, что у пациентов с СН, по мере коррекции отёчного синдрома, наблюдаются снижение времени наступления максимальной концентрации препарата в сыворотке крови (Tmax) на 27 % и увеличение Cmax на 29 %, что может свидетельствовать об уменьшении замедления скорости абсорбции (на 57 %). Поскольку фуросемид в основном выводится с мочой в неизменённом виде, наблюдаемые изменения Cmax и Tmax могли быть связаны с задержкой опорожнения желудка, снижением перистальтики кишечника или отёком стенки кишечника [45].
Vargo DL и соавт. [47] исследовали фармакокинетику фуросемида и торсемида у 16 пациентов с СН II или III ФК по NYHA и ФВ ЛЖ ≤40 %. В открытом исследовании фуросемид вводился в дозе 40 мг перорально и 20 мг внутривенно, а торсемид — в дозе 10 мг перорально и внутривенно. Образцы крови брали до и в течение 36 ч после введения препарата. Было обнаружено, что Foral составляет 89,3 % для торсемида и 71,8 % для фуросемида, исходя из отношения AUC pеr os (величина AUC при пероральном пути введения) к AUCiv (величина AUC при внутривенном пути введения) [47]. Исходя из полученных данных, ФК торасемида при пероральном приёме у пациентов с СН не отличается от таковой у здоровых субъектов, за исключением увеличения Т1/2 и более высоких значений AUC pеr os.
Gottlieb SS и соавт. [46] изучали ФК фуросемида и торсемида у 44 пациентов с СН III или IV ФК по NYHA. Пациентам с выраженными застойными явлениями давали либо фуросемид 20–400 мг, либо торасемид 10–160 мг. Авторы выявили незначительное увеличение многих фармакокинетических параметров фуросемида (например, Cmax, Тmax, AUC per os и Clrenal), а также значительное увеличение Cmax и Tmax для торасемида. Wargo KA и Banta WM [46] в своём обзоре пришли к выводу, что у пациентов с ХСН торасемид и буметанид имеют более высокую абсорбцию при пероральном приёме, чем фуросемид, другие их фармакокинетические параметры изменяются в гораздо меньшей степени, чем у фуросемида. Основываясь на этих данных, авторы cчитают, что торсемид или буметанид, а не фуросемид следует рассматривать как терапию первой линии при лечении отёчного синдрома / застойных явлений у пациентов с СН.
Гидрохлоротиазид ингибирует активную реабсорбцию хлора в кортикальной части восходящей петли Генле. После перорального приёма он абсорбируется из ЖКТ на 60–80 %, а пиковые концентрации в плазме достигаются через 1,5–5 часов [14], продолжительность действия — от 8 до 12 часов. Более 95 % препарата выводится через почки в неизменённом виде [14]. У пациентов с СН фармакокинетика гидрохлоротиазида может изменяться: снижаются абсорбция из ЖКТ (21–37 % от принятой внутрь дозы) и почечный клиренс [44][49]. Таким образом, полнота и скорость всасывания гидрохлоротиазида при его приёме внутрь могут быть крайне вариабельными, особенно у пациентов с тяжёлой ХСН, это может негативно сказаться на эффективности терапии.
В таблице 3 систематизирована информация о потенциальных изменениях фармакокинетики петлевых диуретиков у пациентов с ХСН, полученная из открытых литературных источников [41–49].
Биодоступность дигоксина при приёме внутрь составляет 60–80 %. Инкапсулированный гелевый препарат имеет улучшенную биодоступность — примерно до 90–100 % [14]. Т1/2 дигоксина составляет 24–48 часов. Основной путь выведения — почками с мочой. При почечной недостаточности объём его распределения уменьшается (370 л против 510 л у здоровых людей), а T1/2 удлиняется [14].
Данные о влиянии СН на абсорбцию и другие фармакокинетические параметры дигоксина противоречивы. В некоторых исследованиях [54][55] не выявлено изменений абсорбции дигоксина у пациентов с СН, а в другом исследовании [56] у 2 пациентов с тяжёлой СН по сравнению с пациентами с более лёгкой степенью СН обнаружены клинически значимое увеличение времени достижения пиковой концентрации и снижение Cmax в сыворотке крови. Вероятно, эти изменения обусловлены снижением скорости абсорбции дигоксина за счёт снижения перистальтики ЖКТ вследствие повышения симпатической активности, а также застойными явлениями в стенке кишечника и снижением кровотока в сосудах кишечника [58]. Дигоксин обладает вазоконстрикторными эффектами и может дополнительно снижать кровоток в сосудах брыжейки [59], что ещё больше снижает скорость его всасывания из просвета ЖКТ.
По данным одних исследований, Т1/2 и Сlrenal дигоксина у пациентов с СН не изменяются [54][55], однако в другом исследовании [59] у пациентов с ФП и ХСН было выявлено увеличение равновесной концентрации дигоксина в сыворотке крови и снижение его почечного клиренса (2,88 ± 1,26 против 4,26 ± 2,16 л/ч) по сравнению с пациентами с ФП, но сохранной насосной функцией сердца.
У большинства больных СН нет необходимости корректировать дозу дигоксина, однако при наличии нарушения функции почек его дозу необходимо уменьшить. В идеале, следует проводить регулярный мониторинг концентрации дигоксина в сыворотке крови, что позволит избежать развития НР.
В таблице 4 систематизирована информация о потенциальных изменениях фармакокинетики дигоксина у пациентов с ХСН, полученная из открытых литературных источников [59–63].
Таблица 4
Исследования по изучению фармакокинетики дигоксина у пациентов с СН [59–63]
Table 4
Summary of studies of pharmacokinetics of digoxin in patients with heart failure [59–63]
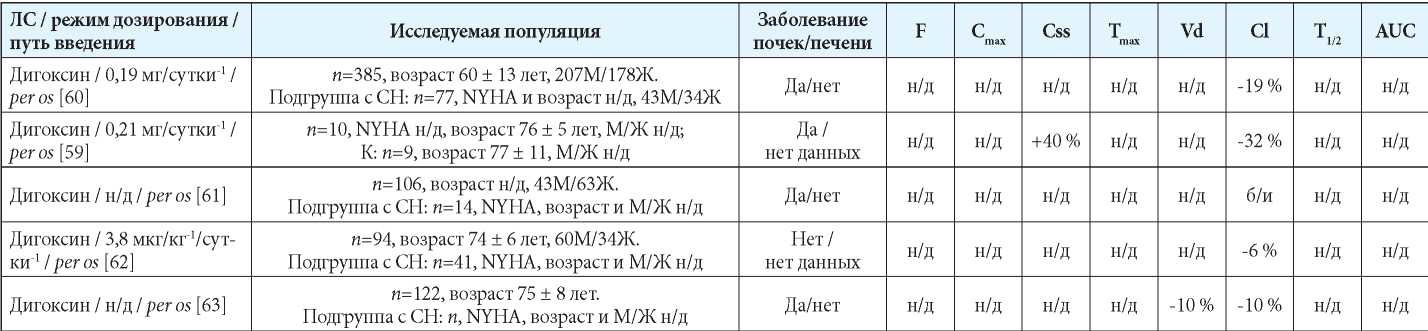
Примечания: б/и — без статистически значимых изменений; в/в — внутривенно; Ж — женщины; К — контроль (группа контроля); М — мужчины; н/д — нет данных; CН — сердечная недостаточность; AUC — area under curve (площадь под кривой); Cmax — максимальная концентрация в сыворотке крови, Cl — клиренс, Сss — равновесная концентрация в сыворотке крови; F — биодоступность; NYHA — New York Heart Association Functional Classification (функциональная классификация сердечной недостаточности Нью-Йоркской ассоциации сердца); Tmax — время достижения максимальной концентрации в сыворотке крови; T1/2 — период полувыведения; Vd — объём распределения.
Notes: б/и — no statistically significant changes; в/в — intravenously; Ж — women; К — control (control group); M — men; н/д — no data; CH — heart failure; AUC — area under curve; Cmax — maximum concentration in blood serum; Cl — clearance, Сss — equilibrium concentration in blood serum, F — bioavailability; NYHA — New York Heart Association Functional Classification; Tmax — time to reach the maximum concentration in the blood serum; T1/2 — half-life; Vd — volume of distribution.
В клинических исследованиях [66–68] о влиянии СН на фармакокинетику цибензолина, дизопирамида и ибутилида не было выявлено статистически значимых различий значений фармакокинетических параметров этих ЛС у пациентов с СН с сохранённой функцией почек и печени по сравнению с контрольной группой лиц [66–68]. Данные о влиянии СН на ФК лидокаина, мексилетина и прокаинамида противоречивы. С одной стороны, по данным ряда исследований [69–71], у пациентов с СН возможно значительное сокращение Vd (33 %), уменьшение Cl (от 36 до 37 %) и значительное удлинение Т1/2 (от +62 до +628 %) лидокаина [69–71]. В других исследованиях [69–71] каких-либо существенных различий в Vd и Т1/2 лидокаина у пациентов с СН по сравнению с контрольной группой не обнаружено [69–71]. По данным одного исследования [72], у пациентов с СН был значительно снижен Cl мексилетина (на 33 %), однако в другом исследовании [73] подобных изменений выявлено не было. В двух исследованиях у пациентов с СН обнаружено значительное снижение Сl прокаинамида (от 17 до 49 %), в одной публикации сообщалось о значительном увеличении (+114 %) Т1/2 прокаинамида, тогда как в других работах статистически значимых изменений фармакокинетических параметров прокаинамида у пациентов с СН не отмечено [74–75]. У пациентов с СН выявлено значительное увеличение Tmax (+127 %) и значительное уменьшение Vd (32 %) и Cl (от 34 до 72 %) хинидина [76], увеличение Vd (+39 %) и Cl (+94 %) никорандила по сравнению с контрольной группой [77], снижение Cmax (28 %), увеличение Tmax (+72 %) и, в меньшей степени, Т1/2 (+29 %) — изосорбида-5-мононитрата [78], значительных изменений биодоступности или Сl изосорбида динитрата не обнаружено [79]. Фармакокинетические параметры БКК нифедипина и антагониста рецепторов эндотелина бозентана у пациентов СН не изменялись [80].
По сравнению с контрольной группой, у пациентов с СН был меньше Сl (58 %) и несколько более длительное Т1/2 (+21 %) эноксимона [81], наблюдались снижение Cl (от 26 до 56 %) адреналина, норадреналина и изопреналина и заметное увеличение стационарных (равновесных) концентраций (от +49 до +204 %) адреналина и норадреналина [82–84]. Фармакокинетика левосимендана у пациентов с СН не подвергалась значительным изменениям [85].
В литературе имеются данные исследований фармакокинетических параметров у пациентов с СН теофиллина, толваптана, дабигатрана, кониваптана, дарбэпоэтина альфа. В исследованиях по фармакокинетике теофиллина у пациентов с СН выявлено значительное снижение его Cl (от 25 до 69 %) [86] и увеличение (от +84 до +267 %) T1/2 теофиллина [87][88]. У пациентов с СН Vd и Cl толваптана были ниже (на 40–49 % и 42–55 % соответственно) по сравнению с контрольной группой. У пациентов с СН, принимавших дабигатран, отмечено небольшое, но при этом статистически значимое снижение его Cl (-7 %) и небольшое увеличение AUC (+7 %) [89]. Статистически значимых изменений фармакокинетики кониваптана и дарбэпоэтина альфа у пациентов с СН не выявлено [90].
Таким образом, у пациентов с ХСН возможно изменение фармакокинетики многих ЛС, применяемых для лечения данной серьёзной и жизнеугрожающей нозологии. Индивидуальный подбор дозы и режима дозирования с учётом особенностей фармакокинетики ЛС у пациентов с ХСН будет способствовать повышению качества жизни и профилактике потенциальных НР.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Conflict of interests. Th e authors declare no conflict of interest.
Благодарности. Работа выполнена без спонсорской поддержки.
Acknowledgements. Th e study was performed without external funding.
Участие авторов. Переверзев А. П. — сбор, анализ и систематизация данных научной литературы, написание текста статьи, оформление, ответственность за все аспекты работы, связанные с достоверностью данных; Остроумова О. Д. — написание текста статьи, критический пересмотр содержания, утверждение окончательного варианта статьи для публикации.
Participation of authors. Pereverzev AP — collection, analysis, and systematisation of literature data, writing and formatting of the paper, carrying responsibility for all aspects of the study related to data reliability; Ostroumova OD — revision of the paper, approval of the final version of the paper for publication.
1. Сычев Д. А., Остроумова О. Д., Кочетков А. И. и др. Лекарственно-индуцированные заболевания: эпидемиология и актуальность проблемы. Фарматека. 2020;27(5):77–84. [Sychev DA, Ostroumova OD, Kochetkov AI et al. Drug-induced diseases: epidemiology and urgency of the problem. Pharmateca. 2020;27(5):77–84. (In Russ).]. doi: 10.18565/pharmateca.2020.5.77-84
2. Tisdale JE, Miller DA. Drug Induced Diseases: Prevention, Detection, and Management. 3rd Ed. Bethesda, Md.: American Society of Health-System Pharmacists; 2018:1399 р.
3. Министерство здравоохранения Российской Федерации. Клинические рекомендации «Хроническая сердечная недостаточность». 2020. ID 156. [Ministry of Health of the Russian Federation. Clinical recommendations “Chronic heart failure”. 2020. ID 156. (In Russ).]. Доступно по: https://cr.minzdrav.gov.ru. Ссылка активна на 23.12.2021.
4. Facts & Comparison. Drug facts and comparisons. Lippincott Williams & Wilkinser Health; 2012. https://www.logobook.ru/prod_show.php?object_uid=12017462 (дата обращения: 29.12.2021).
5. Heel RC, Brogden RN, Speight TM, Avery GS. Captopril: a preliminary review of its pharmacological properties and therapeutic efficacy. Drugs. 1980;20(6):409–52. doi: 10.2165/00003495-198020060-00001
6. Nishida M, Matsuo H, Sano H, Obata H, Yasuda H. Effect of captopril on congestive heart failure. Jpn Circ J. 1990;54(12):1497–502. doi: 10.1253/jcj.54.12_1497
7. Ulm EH, Hichens M, Gomez HJ, et al. Enalapril maleate and a lysine analogue (MK-521): disposition in man. Br J Clin Pharmacol. 1982;14(3):357– 62. doi: 10.1111/j.1365-2125.1982.tb01991.x
8. Schwartz JB, Taylor A, Abernethy D et al. Pharmacokinetics and pharmacodynamics of enalapril in patients with congestive heart failure and patients with hypertension. J Cardiovasc Pharmacol. 1985;7(4):767–76. doi: 10.1097/00005344-198507000-00023
9. Singhvi SM, Duchin KL, Morrison RA et al. Disposition of fosinopril sodium in healthy subjects. Br J Clin Pharmacol. 1988;25(1):9–15. doi: 10.1111/j.1365-2125.1988.tb03275.x
10. Kostis JB, Garland WT, Delaney C et al. Fosinopril: pharmacokinetics and pharmacodynamics in congestive heart failure. Clin Pharmacol Ther. 1995;58(6):660–5. doi: 10.1016/0009-9236(95)90022-5
11. Beermann B. Pharmacokinetics of lisinopril. Am J Med. 1988;85(3B):25– 30. doi: 10.1016/0002-9343(88)90346-4
12. Zestril (lisinopril) tablets [package insert]. Wilmington: AstraZeneca; 2013. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/019777s076lbl.pdf (дата обращения: 29.12.2021).
13. Gautam PC, Vargas E, Lye M. Pharmacokinetics of lisinopril (MK521) in healthy young and elderly subjects and in elderly patients with cardiac failure. J Pharm Pharmacol. 1987;39(11):929–31. doi: 10.1111/j.2042-7158.1987.tb03130.x
14. Государственный реестр лекарственных средств Минздрава России. Available at: https://grls.rosminzdrav.ru/ (дата обращения: 15.10.2021).
15. Bellissant E, Giudicelli JF. Pharmacokinetic-pharmacodynamic model for perindoprilat regional haemodynamic effects in healthy volunteers and in congestive heart failure patients. Br J Clin Pharmacol. 2001;52(1):25–33. doi: 10.1046/j.0306-5251.2001.01410.x
16. Breslin E, Posvar E, Neub M et al. A pharmacodynamic and pharmacokinetic comparison of intravenous quinaprilat and oral quinapril. J Clin Pharmacol. 1996;36(5):414–21. doi: 10.1002/j.1552-4604.1996.tb05028.x
17. Squire IB, Macfadyen RJ, Lees KR et al. Haemodynamic response and pharmacokinetics after the first dose of quinapril in patients with congestive heart failure. Br J Clin Pharmacol. 1994;38(2):117–23. doi: 10.1111/j.1365-2125.1994.tb04334.x
18. Begg EJ, Robson RA, Ikram H et al. The pharmacokinetics of quinapril and quinaprilat in patients with congestive heart failure. Br J Clin Pharmacol. 1994;37(3):302–4. doi: 10.1111/j.1365-2125.1994.tb04280.x
19. Eckert HG, Badian MJ, Gantz D et al. Pharmacokinetics and biotransformation of 2-[N-[(S)-1-ethox-ycarbonyl-3-phenylpropyl]- L-alanyl]-(1S,3S,5S)-2-azabicyclo[3.3.0]octane-3-carboxylic acid (Hoe 498) in rat, dog and man. Arzneimittelforschung. 1984;34(10B):1435–47.
20. Kondo K, Ohashi K, Saruta T et al. Tolerability, pharmacodynamics and -kinetics of Hoe 498 after multiple administration of 5 mg for 15 days in healthy male subjects. Jpn Pharmacol Ther. 1986;14(2):803–23.
21. Anpo Y, Mori S, Yokoi H et al. Pharmacokinetics of candesartan cilexetil (TCV-116) in patients with chronic heart failure. J N Remedies Clin. 1996;45(9):1662–8.
22. Aoi W. Pharmacokinetics study of angiotensin II receptor antagonist (TCV-116) in elderly patients in hypertension [in Japanese]. Rinsho Iyaku. 1996;12(11):2429–41.
23. Lo MW, Goldberg MR, McCrea JB et al. Pharmacokinetics of losartan, an angiotensin II receptor antagonist, and its active metabolite EXP3174 in humans. Clin Pharmacol Ther. 1995;58(6):641–9. doi: 10.1016/0009-9236(95)90020-9
24. Lo MW, Toh J, Emmert SE et al. Pharmacokinetics of intravenous and oral losartan in patients with heart failure. J Clin Pharmacol. 1998;38(6):525– 32. doi: 10.1002/j.1552-4604.1998.tb05790.x
25. De Leon Diaz de Leon, Edgar & Almendra-Pegueros, Rafael & Gonzalez-Correa, Ja & Tejerina, Teresa & Medina, Úrsula & Gordillo-Moscoso, Antonio. Factors associated to therapeutic failure of losartan in patients with heart failure. pharmacokinetic model. The FASEB Journal. 2019. https://www.fasebj.org/doi/10.1096/fasebj.2019.33.1_supplement.680.10 (дата обращения: 29.12.2021). doi: 10.1096/fasebj.2019.33.1_supplement.680.10
26. Brookman LJ, Rolan PE, Benjamin IS et al. Pharmacokinetics of valsartan in patients with liver disease. Clin Pharmacol Ther. 1997;62(3):272–8. doi: 10.1016/S0009-9236(97)90029-1
27. Prasad PP, Yeh CM, Gurrieri P et al. Pharmacokinetics of multiple doses of valsartan in patients with heart failure. J Cardiovasc Pharmacol. 2002;40(5):801–7. doi: 10.1097/00005344-200211000-00018
28. Cyong J-C, Uebaba K. Phase I study of angiotensin II receptor antagonist, CGP 48933 (valsartan)-multiple administration study. Rinsho Iyaku. 1998;14(10):1727–43.
29. Leopold G, Pabst J, Ungethum W, Buhring KU. Basic pharmacokinetics of bisoprolol, a new highly beta 1-selective adrenoceptor antagonist. J Clin Pharmacol. 1986;26(8):616–21. doi: 10.1002/j.1552-4604.1986.tb02959.x
30. Nikolic VN, Jevtovic-Stoimenov T, Velickovic-Radovanovic R, et al. Population pharmacokinetics of bisoprolol in patients with chronic heart failure. Eur J Clin Pharmacol. 2013;69(4):859–65. doi: 10.1007/s00228-012-1427-y
31. Tenero D, Boike S, Boyle D, et al. Steady-state pharmacokinetics of carvedilol and its enantiomers in patients with congestive heart failure. J Clin Pharmacol. 2000;40(8):844–53. doi: 10.1177/00912700022009576
32. Nikolic VN, Jankovic SM, Velickovic-Radovanovic R, et al. Population pharmacokinetics of carvedilol in patients with congestive heart failure. J Pharm Sci. 2013;102(8):2851–8. doi: 10.1002/jps.23626
33. Horiuchi I, Nozawa T, Fujii N, et al. Pharmacokinetics of R- and S-Carvedilol in routinely treated Japanese patients with heart failure. Biol Pharm Bull. 2008;31(5):976–80. doi: 10.1248/bpb.31.976
34. Saito M, Kawana J, Ohno T, et al. Population pharmacokinetics of Rand S-carvedilol in Japanese patients with chronic heart failure. Biol Pharm Bull. 2010;33(8):1378–84. doi: 10.1248/bpb.33.1378
35. Taguchi M, Nozawa T, Mizumaki K, et al. Nonlinear mixed effects model analysis of the pharmacokinetics of metoprolol in routinely treated Japanese patients. Biol Pharm Bull. 2004;27(10):1642–8. doi: 10.1248/bpb.27.1642
36. Laer S, Mir TS, Behn F, et al. Carvedilol therapy in pediatric patients with congestive heart failure: a study investigating clinical and pharmacokinetic parameters. Am Heart J. 2002;143(5):916–22. doi: 10.1067/mhj.2002.121265
37. Lefebvre J, Poirier L, Poirier P, et al. The influence of CYP2D6 phenotype on the clinical response of nebivolol in patients with essential hypertension. Br J Clin Pharmacol. 2006;63(5):575–82. doi: 10.1111/j.1365-2125.2006.02796.x
38. Zannad F, McMurray JJ, Krum H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364(1):11–21. doi: 10.1056/NEJMoa1009492
39. Cook CS, Berry LM, Bible RH, et al. Pharmacokinetics and metabolism of [14C]eplerenone after oral administration to humans. Drug Metab Dispos 2003;31(11):1448–55.
40. Ravis WR, Reid S, Sica DA, Tolbert DS. Pharmacokinetics of eplerenone after single and multiple dosing in subjects with and without renal impairment. J Clin Pharmacol 2005;45(7):810–21.
41. Pitt B, Zannad F. Eplerenone: is it time to add this drug to current heart failure therapy? Ther Adv Chronic Dis. 2012;3(1):5–9. doi: 10.1177/2040622311429891
42. Brater DC, Day B, Burdette A, Anderson S. Bumetanide and furosemide in heart failure. Kidney Int. 1984;26(2):183–9. doi: 10.1038/ki.1984.153
43. Brater DC, Seiwell R, Anderson S, et al. Absorption and disposition of furosemide in congestive heart failure. Kidney Int. 1982;22(2):171–6. doi: 10.1038/ki.1982.149
44. Andreasen F, Mikkelsen E. Distribution, elimination and effect of furosemide in normal subjects and in patients with heart failure. Eur J Clin Pharmacol. 1977;12(1):15–22. doi: 10.1007/BF00561400
45. Vasko MR, Cartwright DB, Knochel JP, et al. Furosemide absorption altered in decompensated congestive heart failure. Ann Intern Med. 1985;102(3):314–8. doi: 10.7326/0003-4819-102-3-314
46. Gottlieb SS, Khatta M, Wentworth D, et al. The effects of diuresis on the pharmacokinetics of the loop diuretics furosemide and torsemide in patients with heart failure. Am J Med. 1998;104(6):533–8. doi: 10.1016/s0002-9343(98)00111-9
47. Vargo DL, Kramer WG, Black PK, et al. Bioavailability, pharmacokinetics, and pharmacodynamics of torsemide and furosemide in patients with congestive heart failure. Clin Pharmacol Ther. 1995;57(6):601–9. doi: 10.1016/0009-9236(95)90222-8
48. Beermann B, Groschinsky-Grind M. Pharmacokinetics of hydrochlorothiazide in man. Eur J Clin Pharmacol. 1977;12(4):297–303. doi: 10.1007/BF00607430
49. Beermann B, Groschinsky-Grind M. Pharmacokinetics of hydrochlorothiazide in patients with congestive heart failure. Br J Clinl Pharmacol. 1979;7(6):579–83. doi: 10.1111/j.1365-2125.1979.tb04646.x
50. Murray MD, Haag KM, Black PK, et al. Variable furosemide absorption and poor predictability of response in elderly patients. Pharmacotherapy. 1997;17(1):98–106.
51. Williams PE, Brown AN, Rajaguru S, et al. The pharmacokinetics and bioavailability of cilazapril in normal man. Br J Clin Pharmacol. 1989;27 Suppl 2(Suppl 2):181S–188S.
52. Wargo KA, Banta WM. A comprehensive review of the loop diuretics: should furosemide be first line? Ann Pharmacother. 2009;43(11):1836–47. doi: 10.1345/aph.1M177
53. Applefeld MM, Adir J, Crouthamel WG, Roffman DS. Digoxin pharmacokinetics in congestive heart failure. J Clin Pharmacol. 1981;21(2):114– 20. doi: 10.1002/j.1552-4604.1981.tb01760.x
54. Ohnhaus EE, Vozeh S, Nuesch E. Absorption of digoxin in severe right heart failure. Eur J Clin Pharmacol. 1979;15(2):115–20. doi: 10.1007/BF00609874
55. Oliver GC, Taxman R, Frederickson R. Influence of congestive heart failure on digoxin level. Proceedings of in Symposium on Digitalis. 1973:336–47.
56. Braunwald W. Heart disease: a textbook of cardiovascular medicine. 2nd ed. W.B. Saunders Company; 1984:648.
57. Ther L, Winne D. Drug absorption. Annu Rev Pharmacol. 1971;11:57– 70. doi: 10.1146/annurev.pa.11.040171.000421
58. Naafs MAB, van der Hoek C, van Duin S, et al. Decreased renal clearance of digoxin in chronic congestive heart failure. Eur J Clin Pharmacol. 1985;28(3):249–52. doi: 10.1007/BF00543318
59. Korhonen UR, Jounela AJ, Pakarinen AJ, et al. Pharmacokinetics of digoxin in patients with acute myocardial infarction. Am J Cardiol. 1979;44(6):1190–4. doi: 10.1016/0002-9149(79)90187-5
60. Yukawa E, Honda T, Ohdo S, et al. Population-based investigation of relative clearance of digoxin in Japanese patients by multiple trough screen analysis: an update. J Clin Pharmacol. 1997;37(2):92–100. doi: 10.1002/j.1552-4604.1997.tb04766.x
61. Yukawa E, Suematu F, Yukawa M, et al. Population pharmacokinetics of digoxin in Japanese patients: a 2-compartment pharmacokinetic model. Clin Pharmacokinet. 2001;40(10):773–81. doi: 10.2165/00003088-200140100-00005
62. Yukawa M, Yukawa E, Suematsu F, et al. Determination of digoxin clearance in Japanese elderly patients for optimization of drug therapy: a population pharmacokinetics analysis using nonlinear mixed-effects modelling. Drugs Aging. 2011;28(10):831–41. doi: 10.2165/11594230-000000000-00000
63. Massarella JW, Silvestri T, DeGrazia F, et al. Effect of congestive heart failure on the pharmacokinetics of cibenzoline. J Clin Pharmacol. 1987;27(3):187–92.
64. Bonde J, Angelo HR, Bodtker S, et al. Kinetics of disopyramide after intravenous infusion to patients with myocardial infarction and heart failure. Acta Pharmacol Toxicol (Copenh). 1985;56(4):278–82. doi: 10.1111/j.1600-0773.1985.tb01290.x
65. Tisdale JE, Overholser BR, Sowinski KM, et al. Pharmacokinetics of ibutilide in patients with heart failure due to left ventricular systolic dysfunction. Pharmacotherapy. 2008;28(12):1461–70. doi: 10.1592/phco.28.12.1461
66. Thomson PD, Melmon KL, Richardson JA, et al. Lidocaine pharmacokinetics in advanced heart failure, liver disease, and renal failure in humans. Ann Intern Med. 1973;78(4):499–508. doi: 10.7326/0003-4819-78-4-499
67. Prescott LF, Adjepon-Yamoah KK, Talbot RG. Impaired lignocaine metabolism in patients with myocardial infarction and cardiac failure. Br Med J. 1976;1(6015):939–41. doi: 10.1136/bmj.1.6015.939
68. Cusson J, Nattel S, Matthews C, et al. Age-dependent lidocaine disposition in patients with acute myocardial infarction. Clin Pharmacol Ther. 1985;37(4):381–6. doi: 10.1038/clpt.1985.58
69. Kobayashi M, Fukumoto K, Ueno K. Effect of congestive heart failure on mexiletine pharmacokinetics in a Japanese population. Biol Pharm Bull. 2006;29(11):2267–9. doi: 10.1248/bpb.29.2267
70. Vozeh S, Katz G, Steiner V, Follath F. Population pharmacokinetic parameters in patients treated with oral mexiletine. Eur J Clin Pharmacol. 1982;23(5):445–51. doi: 10.1007/BF00605996
71. du Souich P, Erill S. Metabolism of procainamide in patients with chronic heart failure, chronic respiratory failure and chronic renal failure. Eur J Clin Pharmacol. 1978;14(1):21–7. doi: 10.1007/BF00560254
72. Reidenberg MM, James M, Dring LG. The rate of procaine hydrolysis in serum of normal subjects and diseased patients. Clin Pharmacol Ther. 1972;13(2):279–84. doi: 10.1002/cpt1972132279
73. Crouthamel WG. The effect of congestive heart failure on quinidine pharmacokinetics. Am Heart J. 1975;90(3):335–9. doi: 10.1016/0002-8703(75)90322-1
74. Iida S, Kinoshita H, Holford NH. Population pharmacokinetic and pharmacodynamic modelling of the effects of nicorandil in the treatment of acute heart failure. Br J Clin Pharmacol. 2008;66(3):352–65. doi: 10.1111/j.1365-2125.2008.03257.x
75. Meissner A, Petersenn S, Heidemann HT, et al. Pharmacokinetics of oral isosorbide-5-mononitrate in patients with ischemic heart failure. Klin Wochenschr. 1991;69(5):213–9. doi: 10.1007/BF01646943
76. Fung HL, Ruggirello D, Stone JA, Parker JO. Effects of disease, route of administration, cigarette smoking, food intake on the pharmacokinetics and circulatory effects of isosorbide dinitrate. Z Kardiol. 1983;72 Suppl. 3:5–10.
77. Chen DG, Feng QP, Wang ZQ, Chen K. Pharmacodynamics and pharmacokinetics of nifedipine in patients with congestive heart failure. Zhongguo Yao Li Xue Bao. 1989;10(3):233–8.
78. Lima JJ, Leier CV, Holtz L, et al. Oral enoximone pharmacokinetics in patients with congestive heart failure. J Clin Pharmacol. 1987;27(9):654–60. doi: 10.1002/j.1552-4604.1987.tb03083.x
79. Kaye DM, Lefkovits J, Cox H, et al. Regional epinephrine kinetics in human heart failure: evidence for extra-adrenal, nonneural release. Am J Physiol. 1995;269(1 Pt 2):H182–8. doi: 10.1152/ajpheart.1995.269.1.H182
80. Stylos L, Parilak L, Green MH, et al. Compartmental analysis of norepinephrine kinetics in congestive heart failure. Am J Cardiol. 1995;75(4):299–301. doi: 10.1016/0002-9149(95)80045-t
81. Leuenberger U, Kenney G, Davis D, et al. Comparison of norepinephrine and isoproterenol clearance in congestive heart failure. Am J Physiol. 1992;263(1 Pt 2):H56–60. doi: 10.1152/ajpheart.1992.263.1.H56
82. Sandell EP, Hayha M, Antila S, et al. Pharmacokinetics of levosimendan in healthy volunteers and patients with congestive heart failure. J Cardiovasc Pharmacol. 1995;26 Suppl 1:S57–62.
83. Powell JR, Vozeh S, Hopewell P, et al. Theophylline disposition in acutely ill hospitalized patients. The effect of smoking, heart failure, severe airway obstruction, and pneumonia. Am Rev Respir Dis. 1978;118(2):229–38. doi: 10.1164/arrd.1978.118.2.229
84. Kuntz HD, Straub H, May B. Theophylline elimination in congestive heart failure. Klin Wochenschr. 1983;61(21):1105–6. doi: 10.1007/BF01496473
85. Cuzzolin L, Schinella M, Tellini U, et al. The effect of sex and cardiac failure on the pharmacokinetics of a slow-release theophylline formulation in the elderly. Pharmacol Res. 1990;22 Suppl 1:137–8. doi: 10.1016/1043-6618(90)90846-6
86. Liesenfeld KH, Lehr T, Dansirikul C, et al. Population pharmacokinetic analysis of the oral thrombin inhibitor dabigatran etexilate in patients with non-valvular atrial fibrillation from the RE-LY trial. J Thromb Haemost. 2011;9(11):2168–75.
87. Mao ZL, Stalker D, Keirns J. Pharmacokinetics of conivaptan hydrochloride, a vasopressin V(1A)/V(2)-receptor antagonist, in patients with euvolemic or hypervolemic hyponatremia and with or without congestive heart failure from a prospective, 4-day open-label study. Clin Ther. 2009;31(7):1542– 50. doi: 10.1016/j.clinthera.2009.07.011
Переверзев Антон Павлович, к. м. н., доцент кафедры терапии и полиморбидной патологии
SPIN-код: 4842-3770
Москва
Остроумова Ольга Дмитриевна, д. м. н., профессор, зав. кафедрой терапии и полиморбидной патологии; профессор кафедры клинической фармакологии и пропедевтики внутренних болезней
SPIN-код: 3910-6585
Москва
Переверзев А.П., Остроумова О.Д. Сердечная недостаточность как фактор риска развития нежелательных реакций. Часть 2: изменение фармакокинетики отдельных лекарственных средств. Качественная клиническая практика. 2021;(4):60-74. https://doi.org/10.37489/2588-0519-2021-4-60-74
Pereverzev A.P., Ostroumova O.D. Heart failure as a risk factor of adverse drug reactions. Part 2: potential changes in pharmacokinetics of some drugs. Kachestvennaya Klinicheskaya Praktika = Good Clinical Practice. 2021;(4):60-74. (In Russ.) https://doi.org/10.37489/2588-0519-2021-4-60-74
НАШИ КНИГИ










Другие журналы
"Издательства ОКИ"
![]()
![]()
![]()
![]()
![]()
ПАРТНЕРЫ
![]()
Адрес редакции и издательства:
ООО «Издательство ОКИ»
115522, Москва, Москворечье ул., 4-5-129
Генеральный директор Афанасьева Елена Владимировна
Тел. + 7 (916) 986-04-65; Email: eva88@list.ru
Обработка персональных данных

































