


Под сертификацией лекарств понимается обязательная оценка и контроль препаратов, что поступают в оборот на территорию РФ. Процедура осуществляется уполномоченным органом, чтобы официально подтвердить их безопасность и соответствие положениям действующих нормативно-правовых актов. Без проведения обязательных оценочных мероприятий предприниматели не вправе выпускать медикаментозные препараты в обращение. Обязательная оценка соответствия лекарствСогласно федеральному закону №323 от 21.11.2011 года, фармацевтические препараты, которые выпускаются в оборот на территории России, подлежат обязательной государственной регистрации. Процедура проводится Минздравом. По итогам предписанных мероприятий оформляется регистрационное удостоверение (РУ). Это официальный документ, который подтверждает, что медикаменты отвечают заявленным техническим и фармацевтическим свойствам, являются безопасными для граждан и окружающей среды при условии использования согласно инструкции производителя и рекомендациям врача. Без получения регистрационного удостоверения предприниматель не вправе начинать производство, реализацию, рекламу медикаментов на территории РФ. В ином случае ему грозят меры административной и уголовной ответственности. Нюансы проведения госрегистрацииСогласно федеральному закону №61 от 12.04.10, регистрация обязательна:
Государственная регистрация не проводится для медикаментов, которые:
Регистрационные процедуры не предусмотрены для изделий одного состава, которые реализуются под разными торговыми наименованиями. Кто проводит регистрацию?Законодательно установлено, что для проведения процедуры вправе обратиться:
После проведения госрегистрации российский изготовитель получает право продавать товар через неограниченное число дистрибьюторов. Регистрационное удостоверение предоставляется заявителю на неограниченный срок. Оно выдается на бумажном носителе, сведения о держателе вносятся в электронный реестр. Получать новый документ компании будет нужно только в том случае, если изменятся рецептуры, инструкции, прилагаемые к препаратам, или регистрационные сведения изготовителя. Как проходит госрегистрация?Порядок организации оценочной процедуры устанавливается Постановлением Правительства №1416 от 27.12.2012. Чтобы оформить разрешительный документ, компания или ИП обращается в Минздрав с заявлением, составленным по унифицированному образцу. В него включаются следующие сведения:
В комплекте с заявлением в ведомство направляется:
Если документы оформлены на иностранном языке, к ним в комплекте предоставляется перевод. Если действует уполномоченное лицо, то требуется доверенность/договор. Заявление в Минздрав подается на бумажном носителе (заявителем, представителем, почтовым отправлением) или по электронным каналам связи. Во втором случае документация заверяется усиленной электронно-цифровой подписью. По законодательству у ведомства имеется 5 дней на проверку полноты и правильности оформления полученных документов. При наличии замечаний заявителю дается 30-ти-дневный срок на их устранение. Если нареканий нет, то за этим следует основная фаза оценки соответствия, которая занимает ориентировочно 50 дней. Итог проведенной работы – выдача предприятию регистрационного удостоверения, которое позволяет начать коммерческую деятельность на территории РФ. Добровольная сертификация лекарствСертификат на лекарства не является обязательным документом для их реализации. Он оформляется по доброй воле изготовителя/ поставщика. Его получение – способ выделить конкурентные преимущества товара в целях увеличения продаж и торговой выручки компании. Начиная сертификацию, предприниматель сам выбирает подтверждаемые характеристики товара. Их нормативные показатели устанавливаются производственным стандартом фирмы. Соответствие проверяется в условиях лаборатории, по итогам испытаний составляется официальный протокол. Сертификат выдается на период, который не может превышать 3-х лет. Как и регистрационное удостоверение, документ действует только на территории России. Он используется держателями как инструмент для маркетингового продвижения товара. |
||
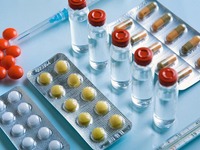
Сертификация лекарственных средств
































НАШИ КНИГИ










Другие журналы
"Издательства ОКИ"
![]()
![]()
![]()
![]()
![]()
ПАРТНЕРЫ
![]()
Popular articles
Keywords
COVID-19
QALY
adverse drug reactions
budget impact analysis
clinical trials
coronavirus
cost-effectiveness analysis
cost-utility analysis
health technology assessment
pharmacoeconomics
pharmacovigilance
анализ влияния на бюджет
анализ затраты-эффективность
анализ минимизации затрат
анализ эффективности затрат
артериальная гипертония
качество жизни
клинические исследования
фармаконадзор
фармакоэкономика
фармакоэпидемиология
121357, Moscow, st. Vatutina, 4, building 2, apt. 22
LLC Publishing House OKI
Tel. +7 (926) 568-17-35
e-mail: clinvest@mail.ru
