


Содержание
Перейти к:
 В. В. Омельяновский,
С. И. Куцев,
П. А. Мухортова,
А. Г. Харитонова,
А. А. Слабикова,
Н. В. Игнатьева,
А. А. Кингшотт,
Т. С. Тепцова,
В. О. Богданова
В. В. Омельяновский,
С. И. Куцев,
П. А. Мухортова,
А. Г. Харитонова,
А. А. Слабикова,
Н. В. Игнатьева,
А. А. Кингшотт,
Т. С. Тепцова,
В. О. Богданова https://doi.org/10.37489/2588-0519-2024-4-82-96
EDN: MQOHEI
Перейти к:
Лекарственные препараты (ЛП) для лечения редких (орфанных) заболеваний характеризуются высокой неопределённостью в отношении их клинической эффективности. В большей степени это обусловлено малым количеством доступных и убедительных клинических исследований и данных по их эффективности. Цель данного обзора заключалась в описании основных проблем, связанных с характеристиками орфанных заболеваний, в т. ч. с характеристиками клинических исследований орфанных ЛП, непосредственно орфанных ЛП, этическими вопросами, а также предложений возможных путей решения данных проблем. В ходе обзора были найдены и описаны основные аспекты, влияющие на диагностику и лечение редких заболеваний, на разработку орфанных ЛП, качество их клинических исследований. Дополнительно были рассмотрены вопросы, касающиеся качества жизни пациентов. Кроме того, предложен ряд организационных и клинических решений по повышению прогнозируемости применения орфанных ЛП, позволяющих добиться большей определённости при их внедрении в практику.
Омельяновский В.В., Куцев С.И., Мухортова П.А., Харитонова А.Г., Слабикова А.А., Игнатьева Н.В., Кингшотт А.А., Тепцова Т.С., Богданова В.О. Пути снижения неопределённости клинической эффективности применения орфанных лекарственных препаратов. Качественная клиническая практика. 2024;(4):82-96. https://doi.org/10.37489/2588-0519-2024-4-82-96. EDN: MQOHEI
Оmelyanovskiy V.V., Kutsev S.I., Mukhortova P.A., Kharitonova A.G., Slabikova A.A., Ignatyeva N.V., Kingshott A.A., Teptsova T.S., Bogdanova V.O. Approaches to reduce the uncertainty in the clinical efficacy of orphan drugs. Kachestvennaya Klinicheskaya Praktika = Good Clinical Practice. 2024;(4):82-96. (In Russ.) https://doi.org/10.37489/2588-0519-2024-4-82-96. EDN: MQOHEI
При общем понимании «орфанных» как относительно редко диагностируемых заболеваний не существует единых стандартов к определению данного термина. Изначально данный термин был применён в США в 1982 г. для описания ситуации отсутствия интереса в разработке лекарственных препаратов (ЛП) для лечения заболеваний с небольшим числом пациентов [1]. В Российской Федерации (РФ) орфанным считается заболевание с распространённостью не более 10 случаев заболевания на 100 тысяч населения, в Европейском Союзе — 1 на 2 тысячи человек, в некоторых странах заявлено суммарное количество случаев заболевания в стране: в Соединенных штатах Америки — не более 200 тысяч случаев, в Японии — не более 50 тысяч случаев [2–5].
В РФ обеспечение пациентов орфанными ЛП финансируется из различных источников. Так, в январе 2021 года Указом Президента РФ был учреждён Фонд поддержки детей с тяжёлыми жизнеугрожающими и хроническими заболеваниями, в том числе редкими (орфанными) заболеваниями «Круг добра», который на данный момент обеспечивает необходимыми ЛП и медицинскими технологиями детей с тяжёлыми жизнеугрожающими, в том числе редкими, заболеваниями [6]. Часть орфанных заболеваний входят в программу высокозатратных нозологий (14 ВЗН), дети с данными нозологиями обеспечиваются препаратами за счёт федерального бюджета через Фонд «Круг добра», взрослые пациенты — за счёт средств федерального бюджета посредством программы 14 ВЗН. Кроме того, пациенты, страдающие заболеваниями, включёнными в «Перечень жизнеугрожающих и хронических прогрессирующих редких (орфанных) заболеваний, приводящих к сокращению продолжительности жизни граждан или их инвалидности», обеспечиваются необходимыми ЛП за счёт средств субъектов РФ по 17 нозологиям [2]. Также возможно обеспечение пациентов орфанными ЛП и в рамках базовой программы обязательного медицинского страхования (ОМС) и в рамках перечня высокотехнологичной медицинской помощи, не включённой в базовую программу ОМС [7].
Держатели бюджета как в РФ, так и за рубежом сталкиваются со схожими проблемами в отношении редких заболеваний. Основной проблемой является неопределённость, прежде всего связанная с результатом лечения, обусловленная малым количеством доступных убедительных данных об эффективности и безопасности лечения.
Ниже представлены основные проблемы, связанные с редкими заболеваниями, проведением клинических исследований орфанных ЛП, этических аспектов их применения, а также возможные варианты их решения (см. рис.).
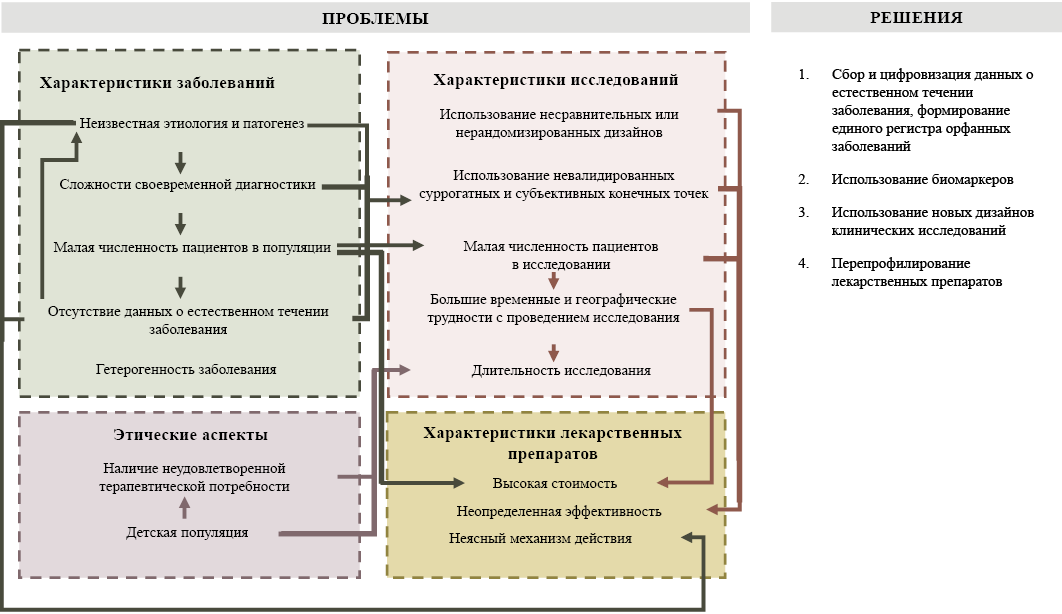
Рис. Характеристики орфанных заболеваний, клинических исследований и лекарственных препаратов, а также методы устранения неопределённости в отношении их эффективности
Fig. Characteristics of orphan diseases, clinical trials and drugs, and methods for addressing uncertainty about their effectiveness
Все основные факторы, определяющие сложности, связанные с орфанными заболеваниями и ЛП, сгруппированы с учётом сферы их возникновения, стрелками обозначены взаимосвязи между различными факторами. Ввиду того, что все рассмотренные факторы оказывают комплексное воздействие на количество и качество данных о клинической эффективности орфанных ЛП и их стоимости, нами был предложен ряд решений, направленных на снижение степени неопределённости клинической эффективности орфанных ЛП.
А) Характеристики заболеваний
Благодаря научному прогрессу, развитию генной и персонализированной медицины и внедрению регуляторных мер поддержки орфанных ЛП, число открытых орфанных заболеваний стремительно увеличивается. Так, в 2020 г. на основании алгоритмического объединения информации из основных источников знаний о редких заболеваниях, было выделено в общей сложности 10 393 редких заболевания [8]. База данных OMIM ежегодно добавляет около 260 новых записей о генетических причинах орфанных заболеваний, база данных Orphanet — около 280 записей (с генетическими мутациями ассоциировано более 70 % орфанных заболеваний) [9][10]. Однако, для более половины выявленных орфанных заболеваний генетические причины возникновения в настоящий момент остаются неизвестными [11]. Так, например, отсутствует единая теория о причинах возникновения бокового амиотрофического склероза [12], идиопатической лёгочной артериальной гипертензии [13] и многих других орфанных заболеваний.
Отсутствие информации об этиологии и патогенезе заболевания не только ограничивает разработку и создание новых молекул с этиопатогенетическим механизмом действия, но и препятствует сбору доказательной базы эффективности и безопасности ЛП в связи с низким уровнем диагностики и крайне маленьким размером популяции пациентов. Не имея достаточной информации о воздействии ЛП на организм, исследования эффективности указанных ЛП ограничиваются использованием невалидированных суррогатных и/или субъективных конечных точек (более подробно данная проблема описана в разделе «Использование невалидированных и субъективных конечных точек в рамках клинических исследований орфанных ЛП»).
Устранить эти пробелы в доказательной базе возможно только при тщательном и своевременном исследовании патологических изменений, способствующих развитию орфанных заболеваний. Для оптимизации поиска на данном этапе может быть внедрено использование алгоритмов машинного обучения, что может помочь не только идентифицировать потенциальные мишени для разработки ЛП, но и оптимизировать разработку новых ЛП для выявленных мишеней [14][15]. Кроме того, выявление молекулярных механизмов, лежащих в основе заболеваний, может способствовать выявлению групп заболеваний с объединёнными молекулярными этиологиями (англ. shared molecular etiologies), например, групп заболеваний, вызванных мутацией в одном гене, заболеваний, вызванных однотипными нарушениями различных генов или заболеваний, вызванных мутациями в разных генах, но затрагивающих единый молекулярный путь [16]. В таком случае разрабатываемые ЛП можно будет таргетировать сразу на несколько заболеваний, проводя, например, корзинные исследования клинической эффективности одной молекулы у пациентов с различными симптомами.
Отсутствие информации об этиологии и патогенезе орфанных заболеваний препятствует своевременной диагностике данных заболеваний. Согласно обзору Adachi T et al., 2023 [17], каждому четвёртому пациенту с орфанным заболеванием в Европейском регионе для постановки диагноза требуется от 5 до 30 лет, в Канаде каждому пятому пациенту требуется до 14 лет, в США пациенту, в среднем, необходимо взаимодействовать с системой здравоохранения 17 раз, в Колумбии — минимум 8 раз. Причинами данной задержки в диагностике, помимо недостатка информации о заболевании, могут быть: недостаточная коммуникация между специалистами, фрагментарность симптомов и гетерогенность заболевания, из-за которого врачи разных специальностей, наблюдающие пациента, не могут корректно диагностировать заболевание.
Общепринятыми методами совершенствования оказания медицинской помощи и улучшения маршрутизации пациентов является разработка клинических рекомендаций и стандартов [18], внедрение дополнительного образования медицинских специалистов [19], создание референтных центров по диагностике и лечению орфанных заболеваний [20]. Помимо данных мер, для оптимизации диагностики пациентов также могут использоваться программы на основе алгоритмов машинного обучения и искусственного интеллекта. В отличие от «традиционных» диагностических исследований, например, секвенирования нового поколения (англ. next generation sequencing) [21], такие методы позволяют при относительно небольших затратах быстро сфокусировать внимание клинического специалиста на нескольких нозологиях и в дальнейшем использовать генетические исследования в качестве подтверждающих, а не скрининговых анализов. Например, кодировщик изображений GestaltMatcher способен сопоставлять портрет определённого пациента с другими пациентами с тем же молекулярным диагнозом [22], сайты-поисковики, такие как https://findzebra.com/1 и https://omim.org/, помогают на основании симптомов и истории (анамнеза) болезни находить информацию о редком заболевании, носителем которого может быть пациент. Такие программы, основанные на алгоритмах поиска и/или искусственном интеллекте, могут способствовать первичному выявлению пациентов и их последующему включению в регистры и клинические исследования, что особенно важно для ультраредких заболеваний, диагностика которых на данный момент затруднена.
Низкая распространённость орфанных заболеваний и проблемы их диагностики приводят к тому, что количество пациентов, страдающих данными заболеваниями, ошибочно считается незначительным для глобальной системы здравоохранения. В то же время, несмотря на ограниченное количество пациентов с отдельным орфанным заболеванием, ввиду большого количества орфанных заболеваний в целом, их кумулятивная распространённость уже сегодня представляется существенной. Так, на основании доступной информации в базе данных Orphanet доля пациентов с такими заболеваниями составляет около 3,5–5,9 % от глобального населения [10], что сопоставимо с распространённостью ишемической болезни сердца у взрослого населения старше 18 лет [23]. Эту проблему усиливают большая длительность процесса постановки диагноза, необходимость дорогостоящих методов диагностики, лечения, а также необходимость междисциплинарного подхода и высокие требования к обучению врачей ввиду высокой гетерогенности клинической картины орфанных заболеваний. Кроме того, определённую роль играют различия в определении орфанных заболеваний в разных государствах и наднациональных организациях.
Малая численность пациентов и сложности с корректной диагностикой орфанных заболеваний приводят к недостатку данных о естественном течении заболевания, в то время как данная информация необходима для разработки и проведения клинических исследований достаточной длительности и с клинически значимыми конечными точками. Чем больше доступно информации о том, как протекает редкое заболевание, тем легче оценить действие некоторых разрабатываемых методов лечения и определить, изменяет ли конкретное лечение течение болезни, влияет ли оно на продолжительность или качество жизни пациентов [24]. Кроме того, необходимо отделять воздействие вмешательства от особенностей течения самого заболевания [25].
Отсутствие данных о естественном течении заболевания и надёжной доказательной базы ЛП может стать препятствием на этапе пострегистрационной оценки технологий здравоохранения (ОТЗ) и включения в систему возмещения. Так, согласно анализу, проведённому бельгийской комиссией по принятию решений о возмещении лекарственных средств при Национальном институте страхования (англ. Commission for Reimbursement of Medicines, проводит оценку досье на возмещение ЛП), заместительная ферментная терапия с использованием аглюкозидазы при болезни Помпе оказалась эффективной при детской форме заболевания, однако доказательная база при «поздней» форме была признана недостаточной [26]. При этом при наличии более полных данных о естественном течении заболевания, возможно было бы более точно ответить на вопрос о добавленной терапевтической ценности ферментной терапии. Именно это создаёт барьеры для внедрения новых орфанных ЛП и определяет необходимость разработки новых подходов ОТЗ именно для орфанных заболеваний.
Даже при условии выявления достаточного количества пациентов и длительного периода наблюдения результаты исследований могут не достигнуть статистической мощности из-за гетерогенности заболевания — наличия внутри одного заболевания фенотипов, имеющих различные клинические проявления. Так, например, выделяют 9 типов мукополисахаридоза [27], каждый из которых характеризуется дефицитом синтеза определённого фермента, связанного с метаболизмом гликозаминогликанов.
В результате, с одной стороны, исследователям в области орфанных заболеваний необходимо набрать большую выборку пациентов с редкими заболеваниями, имеющими сходную клиническую картину, а с другой — высокая детализация исследуемой популяции (например, включение пациентов только с определённым опытом предыдущей терапии) ограничит число включённых пациентов, что снизит статистическую мощность исследования [28]. Более того, вариабельность пациентов внутри групп (например, вариабельность исходных характеристик пациентов) будет приводить к большим значениям дисперсии, чем в классических исследованиях, проводимых на множестве пациентов. Решением данной проблемы может быть использование «новых» дизайнов клинических исследований (см. ниже п. «Использование новых дизайнов клинических исследований»), а также проведение последующих субанализов, рассматривающих более узкую популяцию.
Б) Характеристики клинических исследований орфанных ЛП
Определение дизайна клинического исследования — один из основных вопросов, поднимающихся при разработке ЛП. Планирование исследований орфанных ЛП представляется особенно сложным уже на самом первом этапе — наборе пациентов, что связано с ограниченным количеством больных, часто недостаточным для традиционных методов статистической обработки, а также невозможностью формирования контрольных групп и применения препаратов сравнения (так называемых «активных компараторов»). В связи с этим среди исследований орфанных ЛП чаще встречаются открытые нерандомизированные исследования с небольшим количеством участников, в которых отсутствует активный компаратор или используется несравнительный дизайн [29][30]. Кроме того, распространёнными являются исследования I и II фазы, требующие включения меньшего количества пациентов [29][31], что может быть связано с большим процентом «неуспешных» клинических исследований среди орфанных ЛП.
Отсутствие данных об этиологии, патогенезе и естественном течении редких заболеваний обуславливают частое использование суррогатных (промежуточных) исходов для определения эффективности проводимого лечения. Часто это может быть связано с относительно коротким периодом наблюдения, не позволяющим полноценно оценить конечные исходы новой терапии. Сложность использования суррогатных исходов, отражающих динамику патофизиологических процессов, в большей мере сопряжена с риском отсутствия прямой связи с конечными клинически значимыми исходами. Невалидированные суррогатные точки могут лишь с «определённой вероятностью» устанавливать объективность изменений в клинической картине заболевания, что увеличивает риск некорректной интерпретации полученных результатов [32]. Поэтому применение суррогатных конечных точек связано с необходимостью валидации, то есть предоставления доказательств о влиянии на «конечный» исход [33]. Однако, при оценке заключений о возмещении, рассмотренных Национальным институтом здоровья и клинического совершенствования Великобритании (англ. National Institute for Health and Care Excellence, NICE), и заключений для тех же технологий и показаний, представленных в ОТЗ-агентствах других стран, данные, подтверждающие связь суррогатной точки с конечным исходом, были чётко определены менее, чем в половине случаев [34].
Также в исследованиях орфанных ЛП используются субъективные исходы (шкалы и опросники), что, с одной стороны, позволяет более комплексно оценить влияние ЛП на состояние пациента, с другой — имеет ряд ограничений, связанных с возможным смещением, возникающим при оценке состояния пациента, в т. ч. возникающего из-за отсутствия учёта специфичности проявлений большинства конкретных редких заболеваний, а также небольшого количества пациентов с различными клиническими проявлениями одного заболевания [35][36]. Исходы, сообщаемые пациентами, также, как и исходы, сообщаемые лицами, осуществляющими уход (англ. observer-reported outcomes), и исходы, сообщаемые медицинскими работниками (англ. clinician-reported outcomes), могут быть использованы в качестве дополнительной меры оценки влияния лечения на повседневную жизнь пациента. Однако в ряде случаев при лечении орфанных заболеваний при невозможности использования объективных критериев оценки эффективности ЛП применение данных исходов в качестве основных может считаться оправданным. Так, например, при оценке клинической эффективности трофинетида для лечения синдрома Ретта, основными симптомами которого являются нарушения вербальной коммуникации и ограничения невербальных навыков, в качестве критериев эффективности были использованы исключительно субъективные опросники и шкалы (в том числе созданная специально для оценки нарушений при синдроме Ретта) [37–39]. При регистрации трофинетида было отмечено, что механизм действия препарата при синдроме Ретта остаётся неизвестным, вследствие чего использование субъективных методов позволило дать расширенное понимание степени влияния препарата на качество жизни пациента и лиц, осуществляющих уход [40].
Как было описано ранее, низкая распространённость орфанных заболеваний объясняет достаточно малую численность пациентов, страдающих данными заболеваниями. Исследования новых методов лечения орфанных заболеваний могут быть затруднены, прежде всего, ограниченным количеством пациентов, способных принять участие в том или ином исследовании. Так, в 2015 году Управление по санитарному надзору за качеством пищевых продуктов и лекарственных средств (англ. Food and Drug Administration; FDA) одобрило использование уридина триацетата для лечения наследственной оротической ацидурии на основании клинического исследования, в которое было включено 4 пациента [41], что, на данный момент является наиболее редким заболеванием, для которого есть зарегистрированные опции терапии [42].
Следующим ограничением клинических исследований редких заболеваний является сложность включения пациентов в исследование, обусловленная недостаточной численностью пациентов в популяции и их значительным разбросом (в виде единичных случаев заболевания) по миру.
В публикации Anzelewicz S et al, 2017 г. [43] авторами отмечается ряд опасений, некоторые из которых связанны с тенденцией к глобализации клинических исследований. Например, значительный географический разброс пациентов может замедлить процесс включения пациентов в исследование, а различия в принципах организации медицинской помощи — ограничить условия проведения клинического исследования и увеличить риск получения неточных данных пациентов при проведении клинических исследований «надстранового» уровня.
Географическая удалённость имеет важное значение как в контексте непосредственной физической удаленности от центра проведения клинического исследования, так и ввиду отсутствия специалистов, имеющих опыт ведения таких заболеваний. В случае проживания на значительном расстоянии от центра, для маломобильных и/или пациентов с тяжёлым течением орфанного заболевания частые поездки будут составлять серьёзные трудности, приводя к сокращению частоты посещения осмотров и других диагностических процедур, нарушению методологии сбора данных, и, в конечном итоге, искажению полученных результатов при их оценке. Возможным способом устранения этих препятствий к сбору данных может стать использование цифровых технологий и методов дистанционного мониторинга, которые, помимо расширения географии включения пациентов, позволяют измерять исходы, которые раньше было невозможно зафиксировать (например, ввиду необходимости длительного наблюдения), и улучшить наблюдение за пациентами [44][45].
В контексте длительности проведения исследования могут рассматриваться следующие аспекты. Во-первых, для проведения клинического исследования орфанного ЛП требуется значительно большее время, чем для других ЛП [31]. Эта потребность в основном объясняется описанными выше сложностями включения достаточного количества пациентов, а также широкой географической разбросанностью.
При этом, несмотря на большую длительность проведения исследования, период непосредственного приёма ЛП может быть очень коротким, и среди пациентов это вызывает некоторые опасения в отношении достаточности приёма терапии для проявления клинического эффекта [46]. Так, во время проведения клинического исследования по оценке эффективности терапии эмапалумаба — ЛП, применяемого у пациентов с первичным гемофагоцитарным лимфогистиоцитозом — длительность клинического исследования составила 4 года, в то время как непосредственно приём ЛП осуществлялся, в основном, в течение 8 недель [47][48].
В) Этические аспекты клинических исследований орфанных препаратов
Сегодня почти 70 % орфанных заболеваний манифестируют в детском возрасте [49], при этом нехватка педиатров, участвующих в проведении клинических исследований, сказывается на количестве должного внимания, необходимого для каждого ребёнка, участвующего в исследовании [50]. Так, некоторые исследователи отмечают, что помимо согласия лиц, несущих юридическую ответственность, перед проведением любых манипуляций в рамках клинического исследования необходимо получить согласие несовершеннолетнего в соответствии с его способностями, уважая его желания [51]. Этот фактор усиливается отсутствием достаточного количества педиатров, имеющих опыт в проведении клинических исследований, так как хорошо известно, что именно в педиатрии по сравнению со «взрослой медициной» количество клинических исследований существенно меньше.
Существуют и проблемы с получением согласия на участие в исследовании как со стороны официальных опекунов детей, так и со стороны профессионального сообщества, особенно в случае возможности рандомизации ребёнка с редким заболеванием в плацебо-группу [50]. Кроме того, в некоторых странах, например, в Германии, плацебо-контролируемые клинические исследования с участием несовершеннолетних не допускаются, поскольку требуется, чтобы исследуемый ЛП был показан для лечения симптомов заболевания ребёнка, спасения его жизни, восстановления здоровья или облегчения страданий [52].
Все этические аспекты, касающиеся проведения клинических исследований, в том числе орфанных ЛП, контролируются этическими комитетами. Так, Этический комитет Министерства здравоохранения выносит заключения об этической обоснованности либо необоснованности применения методов профилактики, диагностики, лечения и реабилитации при оказании медицинской помощи в рамках клинической апробации [53], в то время как на базе каждой организации, которая участвует в проведении клинических исследований, создаются также локальные этические комитеты, которые отвечают за защиту прав субъектов исследования и их безопасности при осуществлении научно-исследовательской или медицинской деятельности.
Г) Решения, направленные на устранение неопределённости в отношении эффективности орфанных ЛП
Одним из наиболее комплексных решений по устранению неопределённости в отношении орфанных заболеваний и ЛП, предназначенных для их лечения, является сбор данных естественного течения заболевания. Для орфанных заболеваний такие данные играют важную роль в определении целевой популяции пациентов, текущей практики терапии, распространённости заболевания, оценке клинических исходов и биомаркеров, предикторов прогрессирования заболевания [54][55]. Более того, сбор и агрегация медицинских данных о пациентах, реализованные в виде информационных систем, позволяют автоматически выявлять пациентов с орфанными заболеваниями и формировать из них отдельный набор данных [56].
При наличии качественных данных о естественном течении заболевания, в дальнейшем на их основе возможно будет сформировать группу внешнего контроля для разработанного ЛП. Так, в 2017 году церлипоназа альфа, показанная для лечения позднего детского нейронального цероидного липофусциноза 2-го типа, получила одобрение регулирующих органов на основании результатов нерандомизированного исследования, в котором 22 пациента, получавшие терапию ЛП после появления симптомов заболевания, сравнивались с внешним контролем из 42 пациентов, не получавших лечения (группа естественного течения заболевания). 21 апреля 2017 г. препарат получил положительное заключение Комитета по лекарственным средствам для человека (англ. Committee for Medicinal Products for Human Use; CHMP) Европейского агентства по лекарственным средствам (англ. European Medicines Agency; EMA), а 27 апреля 2017 г. был одобрен в США [57].
Помимо этого, хорошо спланированное исследование естественного течения заболевания помогает выявить пациентов для будущих интервенционных исследований, ускоряя последующий набор за счёт предварительной идентификации лиц, заинтересованных в получении исследуемого продукта, наличия контактов с центрами и врачами, наблюдающими пациентов с данным заболеванием [55].
Для объективизации оценки ответа на исследуемую терапию в качестве суррогатного исхода возможно использование характерных для заболевания биомаркеров [58]. Биомаркер — специфичный индикатор, который может быть объективно измерен и оценен при нормальных биологических или патологических процессах или в качестве фармакологического ответа на терапевтическое вмешательство [59]. Использование данных маркеров может быть полезно при определении необходимой терапевтической дозы или при оценке потенциальных клинически значимых конечных точек на ранней стадии разработки ЛП.
Ярким примером использования биомаркеров в клиническом исследовании орфанных ЛП является зарегистрированный в США деландистроген моксепарвовек — первый ЛП генной терапии для лечения детей в возрасте от 4 лет с миодистрофией Дюшенна [60]. Решение об одобрении было принято на основании предоставленных заявителем данных об увеличении экспрессии белка миодистрофина (суррогатный исход) несмотря на то, что по основному клиническому исходу — улучшение двигательных функций — не было выявлено клинических преимуществ. В данной ситуации регулятором было отмечено, что, с высокой долей вероятности, результаты по данному суррогатному исходу могут предсказывать клиническую эффективность зарегистрированного ЛП [60].
Эффективность исследуемой терапии также может оцениваться посредством измерения метаболитов, преобразующихся в ходе патофизиологических процессов и отражающих активность течения конкретных заболеваний. Так, в рандомизированном контролируемом испытании (РКИ) рекомбинантной L-аспарагиназы по сравнению с нативной L-аспарагиназой у детей с острым лимфобластным лейкозом (ОЛЛ) оценка эффективности проводилась по множеству критериев, основным из которых в контексте определения активности заболевания методом измерения преобразования метаболитов является доля пациентов, достигших снижения уровня аспарагина в сыворотке крови [61]. Аспарагин — основная аминокислота, участвующая в формировании и росте опухолевых клеток при ОЛЛ, однако под действием ЛП L-аспарагиназы происходит разрушение аспарагина, его истощение в сыворотке крови и снижение количества бластов [62]. При этом с учётом вышеописанных особенностей орфанных заболеваний (в т. ч. недостатка данных об этиологии, патогенезе и гетерогенности заболевания), применение метода измерения уровня метаболитов для оценки эффективности ЛП может быть ограничено [63].
В случаях, когда использование традиционных дизайнов клинических исследований невозможно или существенно ограничено (невозможно использование плацебо, нет группы сравнения), решением может стать использование адаптивных дизайнов. Адаптивные дизайны клинических исследований предполагают возможность изменения определённых параметров на основании промежуточных результатов, накапливаемых в ходе исследования, в соответствии с заранее установленными правилами [64]. Помимо адаптивных методов существуют различные менее распространённые виды дизайна (например, N-из-1, дизайн с рандомизированной плацебо-фазой (англ. randomised placebo phase)), которые позволяют максимизировать число пациентов, получающих более эффективное лечение, при относительно малых выборках. Примеры дизайнов клинических исследований, которые могут быть использованы для оценки эффективности орфанных ЛП, приведены в таблице.
Таким образом, применение описанных дизайнов клинических исследований, хотя и сопряжено с необходимостью более тщательного планирования клинических исследований из-за повышенного риска возникновения систематических ошибок, в то же время позволяет получить данные об эффективности лечения в короткие сроки при ограниченном количестве участников.
Таблица
Примеры дизайнов клинических исследований для орфанных лекарственных препаратов
Table
Examples of clinical trial designs for orphan drugs
Название дизайна | Описание дизайна | Преимущества | Недостатки |
Дизайн с адаптивной подгрупповой последовательностью (англ. Adaptive group sequential) | Дизайн, при котором существует несколько групповых последовательных исследования с одним или несколькими промежуточными анализами и заранее определёнными критериями прекращения исследования [65] | — сокращение ожидаемого размера выборки и продолжительности клинических исследований | — существует вероятность необоснованной ранней остановки исследования — выполнение множества статистических тестов без соответствующих поправок может увеличить вероятность возникновения ошибки I рода |
Дизайн с одновременным проведением II и III фазы исследования (англ. Seamless Phase II/III) | Дизайн, при котором выбор наиболее эффективного варианта терапии и/или дозы и сравнительная оценка эффективности с контролем происходит в рамках одного и того же исследования [66]. Данные об исходах обобщаются из двух фаз исследования [67] | — уменьшение времени на проведение исследования — сокращение организационных затрат на организацию исследования | — выбор наиболее эффективной опции терапии на первом этапе может быть основан только на краткосрочных исходах — между первым и вторым этапом необходима пауза для обработки результатов |
Дизайн с пересмотром объёма выборки (англ. sample size re-estimation) | Дизайн, при котором предполагается увеличение (очень редко — уменьшение) размера выборки на основании промежуточных результатов исследования [68] | — повышение вероятности получения статистически значимых результатов | — при включении большего количества пациентов возникает риск подвергнуть большее количество пациентов, чем необходимо, потенциально некачественному лечению — необходимы дополнительные логистические и организационные мероприятия для повторного набора пациентов |
N-из-1 (англ. N-of-1) | Дизайн, при котором единственный пациент, включённый в исследование, получает несколько последовательных видов лечения [69] | — могут быть проведены врачом в рамках рутинной практики — особенности дизайна исследования могут быть подстроены под конкретного пациента | — ограниченная внешняя валидность — не могут быть применены к острым состояниям, необходимо, чтобы заболевание оставалось относительно постоянным в течение всего периода исследования |
Дизайн с рандомизированной плацебо-фазой (англ. Randomised placebo phase) | Дизайн, при котором после фиксированной по длительности фазы плацебо все пациенты переходят на активный ЛП без нарушения ослепления [70][71] | — все пациенты гарантированно получат активный ЛП | — сложность определения длительности периода плацебо и исследования в целом, необходимого для выявления эффективности ЛП — применим в большей степени для высокоэффективных ЛП, которые приводят к достижению положительного ответа в течение короткого промежутка времени |
Дизайн исследования с отсроченным стартом (англ. Delayed start design) | Дизайн, при котором одна группа, ранее принимавшая плацебо, отсрочено будет получать исследуемый ЛП [72][73] | — сокращение длительности терапии препаратом плацебо | — используется для ЛП, обладающих медленным клиническим эффектом, развивающимся в течение времени — меньшее время терапии плацебо снизит разницу в эффектах между группами и потребует увеличения выборки |
Дизайн с рандомизацией после исключения (англ. Randomised withdrawal) | Дизайн, при котором дальнейшая терапия (включая приём плацебо) проводится только тем пациентам, которые продемонстрировали ответ на лечение [73][74] | — оценка эффективности и оптимальной длительности терапии для конкретного пациента, ответившего на лечение | — этические сложности обоснования исключения из исследования — сложность определения критериев сохранения заболевания, поскольку до рандомизации всем пациентам назначается активный ЛП |
Решением проблемы ограничения разработки и создания новых молекул с этиопатогенетическим механизмом действия из-за отсутствия информации об этиологии и патогенезе заболевания может являться перепрофилирование существующих ЛП, что уже отлично работает в индустрии орфанных ЛП. Также перепрофилирование может сократить время разработки и клинических исследований, что позволит быстрее закрыть неудовлетворённые медицинские потребности. Те ЛП, которые не были одобрены по причине недостаточной эффективности по одному показанию, а также ЛП, уже применяющиеся при других нозологиях, при достижении необходимого соотношения пользы и риска могут быть перепрофилированы для лечения других заболеваний [75]. Примерами таких ЛП, одобренными FDA, являются целекоксиб (орфанное показание: семейный аденоматозный полипоз), силденафил и илопрост (лёгочная гипертензия), эверолимус (туберозный склероз), талидомид (множественная миелома) и другие [76, 77].
Обеспечение пациентов с орфанными заболеваниями качественной медицинской помощью представляет собой одну из сложнейших задач, стоящих перед системой здравоохранения. Подходы к организации помощи данным пациентам требует одновременно учитывать целый спектр ограничений — начиная от особенностей самих заболеваний, трудностей проведения клинических исследований, низкого качества данных об эффективности и безопасности возможной терапии, заканчивая социальными медицинскими и этическими аспектами принятия решений.
Описанные в рамках данной статьи ограничения разработки и экспертизы орфанных заболеваний обуславливают высокий уровень неопределённости, стоящий перед всеми участниками этого процесса — индустрией, регуляторами, плательщиком и врачом. Предложенные организационные и клинические подходы направлены на снижение неопределённости при внедрении орфанных технологий в систему здравоохранения, при этом для их имплементации необходимы дальнейшие скоординированные действия со стороны исследовательского, медицинского и пациентского сообществ.
ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ | ADDITIONAL INFORMATION | |
Конфликт интересов Авторы заявляют об отсутствии конфликта интересов. | Conflict of interests The authors state that there is no conflict of interest. | |
Участие авторов Омельяновский В. В. — определение концепции публикации, редактирование текста; Куцев С. И. — редактирование текста; Мухортова П. А. — обзор публикаций по теме статьи (проблемы клинических исследований орфанных ЛП и их решения), написание соответствующих разделов текста, редактирование текста, подготовка рукописи для публикации; Харитонова А. Г. — обзор публикаций по теме статьи (этические проблемы и их решения), написание соответствующих разделов текста, редактирование текста, подготовка рукописи для публикации; Слабикова А. А. — обзор публикаций по теме статьи (проблемы орфанных заболеваний и их решения), написание соответствующих разделов текста, редактирование текста; Игнатьева Н. В. — редактирование текста; Кингшотт А. А. — составление резюме, редактирование текста; Тепцова Т. С. — составление заключения, редактирование текста; Богданова В. О. — редактирование текста. | Authors’ participation Omelyanovsky VV — definition of the publication concept, text editing; Kutsev SI — text editing; Mukhortova PA — review of publications on the topic of the article (problems of clinical trials of orphan drugs and their solutions), writing the relevant sections of the text, text editing, preparing the manuscript for publication; Kharitonova AG — review of publications on the topic of the article (financial and ethical problems and their solutions), writing the relevant sections of the text, text editing, preparing the manuscript for publication; Slabikova AA — review of publications on the topic of the article (problems of orphan diseases and their solutions), writing the relevant sections of the text, text editing; Ignatyeva NV — text editing; Kingshott AA — writing a summary, editing the text; Teptsova TS — writing a conclusion, editing the text; Bogdanova VO — text editing. | |
Финансирование Поисково-аналитическая работа проведена на личные средства авторского коллектива. | Funding The search and analytical work were carried out using the personal funds of the authors’ team. |
1. Название образовано от поговорки «When you hear hoofbeats, think of horses not zebras» — «Когда Вы слышите звук копыт, думайте о лошадях, а не о зебре», означающей, что большинство симптомов являются проявлениями типичных заболеваний. Лошадям, при этом, противопоставляются зебры — пациенты с орфанными заболеваниями, с необычной клинической картиной, которые часто остаются не диагностированными.
1. How Orphan Drugs Came to Be Called “Orphan” [Электронный ресурс] // FDA Law Blog. URL: https://www.thefdalawblog.com/2015/02/how-orphan-drugs-came-to-be-called-orphan/ (Дата обращения: 28.09.2023).
2. Федеральный закон от 21.11.2011 N 323-ФЗ (ред. от 28.12.2022) “Об основах охраны здоровья граждан в Российской Федерации”. [Federal Law of Russian Federation of 21 November 2011 №323-FZ. “Ob osnovakh okhrany zdorov'ya grazhdan v Rossijskoj FederaciI”. (In Russ).] Доступно по: https://minzdrav.gov.ru/documents/7025-federalnyy-zakon-323-fz-ot-21-noyabrya-2011-g. Ссылка активна на 18.03.2024.
3. Rare diseases [Электронный ресурс]. URL: https://research-and-innovation.ec.europa.eu/research-area/health/rare-diseases_en (Дата обращения: 04.09.2023).
4. Rare Diseases at FDA [Электронный ресурс] // FDA, 2022. URL: https:// www.fda.gov/patients/rare-diseases-fda (Дата обращения: 04.09.2023).
5. Overview of Orphan Drug/Medical Device Designation System [Электронный ресурс]. URL: https://www.mhlw.go.jp/english/po-licy/health-medical/pharmaceuticals/orphan_drug.html (Дата обращения: 03.06.2024).
6. Указ Президента РФ от 05.01.2021 № 16 "О создании Фонда поддержки детей с тяжелыми жизнеугрожающими и хроническими заболеваниями, в том числе редкими (орфанными) заболеваниями, “Круг добра”. Доступно по: http://publication.pravo.gov.ru/Document/View/0001202101060001. Ссылка активна на 18.03.2024.
7. Постановление Правительства РФ от 28 декабря 2023 г. № 2353 “О Программе государственных гарантий бесплатного оказания гражданам медицинской помощи на 2024 год и на плановый период 2025 и 2026 годов”. Доступно по: http://static.government.ru/media/files/vB0TvgWlcYbdAUFJomenUk3B0sjTuLA8.pdf. Ссылка активна на 18.03.2024
8. Haendel M, Vasilevsky N, Unni D, et al. How many rare diseases are there? Nat Rev Drug Discov. 2020;19(2):77–78. doi: https://doi.org/10.1038/d41573-019-00180-y.
9. Boycott KM, Rath A, Chong JX, et al. International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. Am J Hum Genet. 2017;100(5):695–705. doi: https://doi.org/10.1016/j.ajhg.2017.04.003.
10. Nguengang Wakap S, Lambert DM, Olry A, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database: 2. Eur J Hum Genet. Nature Publishing Group, 2020;28(2):165–173. doi: https://doi.org/10.1038/s41431-019-0508-0.
11. Greene D, Pirri D, Frudd K, et al. Genetic association analysis of 77,539 genomes reveals rare disease etiologies: 3. Nat Med. Nature Publishing Group, 2023;29(3):679–688. doi: https://doi.org/10.1038/s41591-023-02211-z.
12. Brotman RG, Moreno-Escobar MC, Joseph J, et al. Amyotrophic Lateral Sclerosis // StatPearls. Treasure Island (FL): StatPearls Publishing, 2023.
13. Pahal P, Sharma S. Idiopathic Pulmonary Artery Hypertension // StatPearls. Treasure Island (FL): StatPearls Publishing, 2023.
14. Schaefer J, Lehne M, Schepers J, et al. The use of machine learning in rare diseases: a scoping review. Orphanet Journal of Rare Diseases. 2020;15(1):145. doi: https://doi.org/10.1186/s13023-020-01424-6.
15. Zhavoronkov A, Ivanenkov YA, Aliper A, et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol. 2019;37(9):1038–1040. doi: https://doi.org/10.1038/s41587-019-0224-x.
16. Zanello G, Garrido-Estepa M, Crespo A, et al. Targeting shared molecular etiologies to accelerate drug development for rare diseases. EMBO Molecular Medicine. 2023;15(7):e17159. doi: https://doi.org/10.15252/emmm.202217159.
17. Adachi T, El-Hattab AW, Jain R, et al. Enhancing Equitable Access to Rare Disease Diagnosis and Treatment around the World: A Review of Evidence, Policies, and Challenges. Int J Environ Res Public Health. 2023;20(6):4732. doi: https://doi.org/10.3390/ijerph20064732.
18. Воробьев П.А. Редкие заболевания у взрослых. Проблемы стандартизации в здравоохранении. 2016;3–4:3–9. [Vorobyov PA. Redkie zabolevaniya u vzroslykh. Problemy standartizacii v zdravookhranenii. 2016;3-4:3-9 (In Russ)].
19. Groft SC, Gopal-Srivastava R, Dellon E., et al. How to Advance Research, Education, and Training in the Study of Rare Diseases. Gastroenterology. 2019;157(4):917–921. doi: https://doi.org/10.1053/j.gastro.2019.08.010.
20. Legrand MA, Bagouet F, Merle B, et al. Value of rare diseases reference centers: impact on diagnosis and access to specialized care in fibrous dysplasia of bone. European Journal of Medical Genetics. 2023;66(11):104849. doi: https://doi.org/10.1016/j.ejmg.2023.104849.
21. Vinkšel M, Writzl K, Maver A, et al. Improving diagnostics of rare genetic diseases with NGS approaches. J Community Genet. 2021;12(2):247– 256. doi: https://doi.org/10.1007/s12687-020-00500-5.
22. Hsieh T-C, Bar-Haim A, Moosa S, et al. GestaltMatcher facilitates rare disease matching using facial phenotype descriptors. Nature Genetics. 2022;54(3):349–357. doi: https://doi.org/10.1038/s41588-021-01010-x.
23. Heart Disease Prevalence - Health, United States [Электронный ресурс]. 2023. URL: https://www.cdc.gov/nchs/hus/topics/heart-disease-prevalence.htm (Дата обращения: 04.09.2023).
24. Gavin P. The importance of natural histories for rare diseases. Expert Opinion on Orphan Drugs. Taylor & Francis, 2015;3(8):855–857. doi: https://doi.org/10.1517/21678707.2015.1063415.
25. Garbade SF, Zielonka M, Komatsuzaki S, et al. Quantitative retrospective natural history modeling for orphan drug development. J Inherit Metab Dis. 2021;44(1):99–109. doi: https://doi.org/10.1002/jimd.12304.
26. Dupont AG, Van Wilder PB. Access to orphan drugs despite poor quality of clinical evidence. British Journal of Clinical Pharmacology. 2011;71(4):488–496. doi: https://doi.org/10.1111/j.1365-2125.2010.03877.x.
27. Mucopolysaccharidoses. Diagnostic Imaging: Pediatrics (Third Edition) / ed. Merrow A.C. et al. Elsevier. 2017:950–953. doi: https://doi.org/10.1016/B978-0-323-44306-7.50335-2.
28. Crisafulli S, Sultana J, Ingrasciotta Y, et al. Role of healthcare databases and registries for surveillance of orphan drugs in the real-world setting: the Italian case study. Expert Opinion on Drug Safety. 2019;18(6):497–509. doi: https://doi.org/10.1080/14740338.2019.1614165.
29. Bell SA, Tudur Smith C. A comparison of interventional clinical trials in rare versus non-rare diseases: an analysis of ClinicalTrials.gov. Orphanet J Rare Dis. 2014;9(1):170. doi: https://doi.org/10.1186/s13023-014-0170-0.
30. Logviss K, Krievins D, Purvina S. Characteristics of clinical trials in rare vs. common diseases: A register-based Latvian study. PLoS ONE / ed. Rosenkranz G. 2018;13(4):e0194494. doi: https://doi.org/10.1371/journal.pone.0194494.
31. Jayasundara K, Hollis A, Krahn M, et al. Estimating the clinical cost of drug development for orphan versus non-orphan drugs. Orphanet J Rare Dis. 2019;14(1):12. doi: https://doi.org/10.1186/s13023-018-0990-4.
32. Fleming TR, Powers JH. Biomarkers and surrogate endpoints in clinical trials. Statistics in Medicine. 2012;31(25):2973–2984. doi: https://doi.org/10.1002/sim.5403.
33. Ou F-S, Michiels S, Shyr Y, et al. Biomarker Discovery and Validation: Statistical Considerations. Journal of Thoracic Oncology. 2021;16(4):537– 545. doi: https://doi.org/10.1016/j.jtho.2021.01.1616.
34. Ciani O, Grigore B, Blommestein H, et al. Validity of Surrogate Endpoints and Their Impact on Coverage Recommendations: A Retrospective Analysis across International Health Technology Assessment Agencies. Med Decis Making. 2021;41(4):439–452. doi: https://doi.org/10.1177/0272989X21994553.
35. Feng J, Gao Z, Shi Z, et al. Patient-reported outcomes in Gaucher’s disease: a systematic review. Orphanet J Rare Dis. 2023;18(1):244. doi: https://doi.org/10.1186/s13023-023-02844-w.
36. Slade A, Isa F, Kyte D, et al. Patient reported outcome measures in rare diseases: a narrative review. Orphanet J Rare Dis. 2018;13(1):61. doi: https://doi.org/10.1186/s13023-018-0810-x.
37. Neul JL, Percy AK, Benke TA, et al. Trofinetide for the treatment of Rett syndrome: a randomized phase 3 study. Nat Med. 2023;29(6):1468–1475. doi: https://doi.org/10.1038/s41591-023-02398-1.
38. Percy A, Ryther R, Marsh E, et al. Trofinetide for the treatment of Rett syndrome: an open-label study in girls 2 to 4 years of age (P13-9.005). Neurology. Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology, 2023;100(17),Supplement 2. doi: https://doi.org/10.1212/WNL.0000000000201897.
39. Clinical Review. Информация о лекарственном препарате, размещенная на сайте Управления по контролю качества пищевых продуктов и лекарственных средств США (англ. Food and Drug Administration, FDA) [Электронный ресурс]. URL: https://www.access-data.fda.gov/drugsatfda_docs/nda/2023/217026Orig1s000MedR.pdf. Дата обращения: 30.07.2023
40. DAYBUETM (trofinetide) oral solution [Электронный ресурс]. URL: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/217026s000lbl.pdf. Дата обращения: 30.07.2023
41. Drug Trials Snapshots: XURIDEN [Электронный ресурс]. URL: https://www.fda.gov/drugs/drug-approvals-and-databases/drug-trials-snapshots-xuriden. Дата обращения: 30.07.2023
42. Newton W. Drug development for ultra-rare diseases: What happens when N=1? [Электронный ресурс] // Clinical Trials Arena. 2022. URL: https://www.clinicaltrialsarena.com/features/drug-development-for-ultra-rare-diseases-what-happens-when-n1/ (Дата обращения: 27.07.2023).
43. Anzelewicz S, Garnier H, Rangaswami A, et al. Cultural, geographical and ethical questions when looking to enroll pediatric patients in rare disease clinical trials. Expert Opinion on Orphan Drugs. 2017;5(8):613– 621. doi: https://doi.org/10.1080/21678707.2017.1348293.
44. Bertha A, Alaj R, Bousnina I, et al. Incorporating digitally derived endpoints within clinical development programs by leveraging prior work. NPJ Digit Med. 2023 Aug 10;6(1):139. doi: 10.1038/s41746-023-00886-9.
45. Bill Byrom P. Utilizing DHTs for Clinical Trial Endpoints. Applied Clinical Trials. 2021;30(5).
46. Gaasterland CMW, van der Weide MCJ, du Prie – Olthof MJ, et al. The patient’s view on rare disease trial design – a qualitative study. Orphanet J Rare Dis. 2019;14(1):31. doi: https://doi.org/10.1186/s13023-019-1002-z.
47. Locatelli F, Jordan MB, Allen C, et al. Emapalumab in Children with Primary Hemophagocytic Lymphohistiocytosis. New England Journal of Medicine. 2020;382(19):1811–1822. doi: https://doi.org/10.1056/NEJMoa1911326.
48. 48. Multicentre Study to Assess Safety Tolerability Pharmacokinetics and Efficacy of i.v. Administrations of NI-0501 an Anti-IFNγ mAb in Paediatric Patients With Primary Haemophagocytic Lymphohistiocytosis: Clinical trial registration NCT01818492. clinicaltrials.gov, 2023.
49. Орфанная настороженность при диспансеризации детей. Педиатрическая фармакология. 2021;18(2):156-157.
50. Auvin S, Avbersek A, Bast T, et al. Drug Development for Rare Paediatric Epilepsies: Current State and Future Directions. Drugs. 2019;79(18):1917– 1935. doi: https://doi.org/10.1007/s40265-019-01223-9.
51. Alfonso Farnós I, Alcalde Bezhold G. Clinical research in rare diseases: New challenges, opportunities and ethical issues. An Pediatr (Engl Ed). 2020;93(4):219–221. doi: https://doi.org/10.1016/j.anpede.2020.06.001.
52. Hasford J, Koch A. Erratum to: Ethical aspects of clinical trials in rare diseases. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 2017 Aug;60(8):893. English. doi: 10.1007/s00103-017-2588-8. Erratum for: Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 2017 May;60(5):556-562. doi: 10.1007/s00103-017-2537-6.
53. Приказ Министерства здравоохранения РФ от 10 июля 2015 г. N 435н “Об Этическом комитете Министерства здравоохранения Российской Федерации”. Доступно по: https://minzdrav.gov.ru/documents/9209-prikaz-ministerstva-zdravoohraneniya-rf-ot-10-iyulya-2015-g-435n-ob-eticheskom-komitete-ministerstva-zdravoohraneniya-rossiyskoy-federatsii. Ссылка активна на 18.03.2024.
54. Rare Diseases: Natural History Studies for Drug Development. Draft Guidance for Industry [Электронный ресурс]. 2019. URL: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/rare-diseases-natural-history-studies-drug-development (Дата обращения: 01.12.2023).
55. Bratton E, Holt C, Kromrey S, et al. Natural History Studies for Rare Diseases: Development Strategies for External Comparator Arms Leveraging Real World Insights [Электронный ресурс]. URL: https://www.iqvia.com/library/white-papers/natural-history-studies-for-rare-diseases (Дата обращения: 01.12.2023).
56. Как «Геном Эксперт» комплексно решает задачи генетиков? [Электронный ресурс]. 2023. URL: https://bars.group/press-center/meropriyatiya/kak-genom-ekspert-kompleksno-reshaet-zadachi-genetikov/ (Дата обращения: 30.11.2023).
57. FDA approves first treatment for a form of Batten disease [Электронный ресурс] // FDA. 2020. URL: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-form-batten-disease (Дата обращения: 15.08.2023).
58. Pizzamiglio C, Vernon HJ, Hanna MG, et al. Designing clinical trials for rare diseases: unique challenges and opportunities. Nat Rev Methods Primers. 2022;2(1):s43586-022-00100–00102. https://doi.org/10.1038/s43586-022-00100-2.
59. Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001 Mar;69(3):89-95. doi: 10.1067/mcp.2001.113989.
60. FDA Approves First Gene Therapy for Treatment of Certain Patients with Duchenne Muscular Dystrophy [Электронный ресурс] // FDA. 2023. URL: https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapy-treatment-certain-patients-duchenne-muscular-dystrophy (Дата обращения: 18.08.2023).
61. van der Sluis IM, de Groot-Kruseman H, te Loo M, et al. Efficacy and safety of recombinant E. coli asparaginase in children with previously untreated acute lymphoblastic leukemia: A randomized multicenter study of the Dutch Childhood Oncology Group. Pediatr Blood Cancer. 2018;65(8):e27083. doi: https://doi.org/10.1002/pbc.27083.
62. Jiang J, Batra S, Zhang J. Asparagine: A Metabolite to Be Targeted in Cancers. Metabolites. 2021;11(6):402. doi: https://doi.org/10.3390/metabo11060402.
63. Shah KK, Kogut S, Slitt A. Challenges in Evaluating Safety and Efficacy in Drug Development for Rare Diseases: A Review for Pharmacists. J Pharm Pract. 2021;34(3):472–479. doi: https://doi.org/10.1177/0897190020930972.
64. Pallmann P, Bedding AW, Choodari-Oskooei B, et al. Adaptive designs in clinical trials: why use them, and how to run and report them. BMC Medicine. 2018;16(1):29. doi: https://doi.org/10.1186/s12916-018-1017-7.
65. Adaptive Design Clinical Trials for Drugs and Biologics Guidance for Industry [Электронный ресурс]. FDA, 2020. URL: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/adaptive-design-clinical-trials-drugs-and-biologics-guidance-industry (Дата обращения: 11.09.2023).
66. Stallard N, Todd S. Seamless phase II/III designs. Stat Methods Med Res. 2011;20(6):623–634. doi: https://doi.org/10.1177/0962280210379035.
67. Chow S-C, Chang M. Adaptive design methods in clinical trials – a review. Orphanet J Rare Dis. 2008;3:11. doi: https://doi.org/10.1186/1750-1172-3-11.
68. Collette L. Sample Size Re-Estimation as an Adaptive Design. Applied Clinical Trials. 2021;30(9).
69. Introduction to N-of-1 Trials: Indications and Barriers (Chapter 1) | Effective Health Care (EHC) Program [Электронный ресурс]. URL: https://effectivehealthcare.ahrq.gov/products/n-1-trials/research-20144#toc-1 (Дата обращения: 12.09.2023).
70. Feldman B, Wang E, Willan A, Szalai JP. The randomized placebo-phase design for clinical trials. J Clin Epidemiol. 2001 Jun;54(6):550-7. doi: 10.1016/s0895-4356(00)00357-7.
71. Abrahamyan L, Feldman BM, Tomlinson G, et al. Alternative designs for clinical trials in rare diseases. American Journal of Medical genetics. Part C, Seminars in Medical Genetics. 2016 Dec;172(4):313-331. DOI: 10.1002/ajmg.c.31533.
72. Spineli LM, Jenz E, Großhennig A, et al. Critical appraisal of arguments for the delayed-start design proposed as alternative to the parallel-group randomized clinical trial design in the field of rare disease. Orphanet J Rare Dis. 2017;12(1):140. doi: https://doi.org/10.1186/s13023-017-0692-3.
73. Cornu C, Kassai B, Fisch R, et al. Experimental designs for small randomised clinical trials: an algorithm for choice. Orphanet J Rare Dis. 2013;8(1):48. doi: https://doi.org/10.1186/1750-1172-8-48.
74. Baiardi P, Giaquinto C, Girotto S et al. Innovative study design for paediatric clinical trials. Eur J Clin Pharmacol. 2011;67(S1):109–115. doi: https://doi.org/10.1007/s00228-011-0990-y.
75. Fetro C, Scherman D. Drug repurposing in rare diseases: Myths and reality. Therapie. 2020;75(2):157–160. doi: https://doi.org/10.1016/j.therap.2020.02.006.
76. Rana P, Chawla S. Orphan drugs: trends and issues in drug development. J Basic Clin Physiol Pharmacol. 2018;29(5):437–446. doi: https://doi.org/10.1515/jbcpp-2017-0206.
77. Колбин А.С., Гапешин Р.А., Малышев С.М. Современные проблемы обеспечения орфанными лекарственными средствами и пути их решения. Вопросы современной педиатрии. 2016;15(4):344-351. https://doi.org/10.15690/vsp.v15i4.1584
Омельяновский Виталий Владимирович — д. м. н., профессор; генеральный директор; зав. кафедрой организации здравоохранения и общественного здоровья с курсом оценки технологий здравоохранения
Москва
Куцев Сергей Иванович — д. м. н., профессор, академик РАН, директор
Москва
Мухортова Полина Алексеевна — главный специалист отдела методологического обеспечения проведения комплексной оценки технологий в здравоохранении
Москва
Харитонова Анна Геннадиевна — ведущий специалист отдела методологического обеспечения проведения комплексной оценки технологий в здравоохранении
Москва
Слабикова Александра Алексеевна — ведущий специалист отдела методологического обеспечения проведения комплексной оценки технологий в здравоохранении
Москва
Игнатьева Нелли Валентиновна — к. фарм. н., доцент
Москва
Кингшотт Анастасия Александровна — начальник отдела методологического обеспечения проведения комплексной оценки технологий в здравоохранении; ассистент кафедры организации здравоохранения и общественного здоровья с курсом оценки технологий здравоохранения
Москва
Тепцова Татьяна Сергеевна — зам. начальника отдела методологического обеспечения проведения комплексной оценки технологий в здравоохранении; ассистент кафедры организации здравоохранения и общественного здоровья с курсом оценки технологий здравоохранения
Москва
Богданова Валерия Олеговна — к. м. н., главный специалист отдела организационно-методического обеспечения поддержки деятельности национальных медицинских исследовательских центров; доцент кафедры организации здравоохранения и общественного здоровья с курсом оценки технологий здравоохранения
Москва
Омельяновский В.В., Куцев С.И., Мухортова П.А., Харитонова А.Г., Слабикова А.А., Игнатьева Н.В., Кингшотт А.А., Тепцова Т.С., Богданова В.О. Пути снижения неопределённости клинической эффективности применения орфанных лекарственных препаратов. Качественная клиническая практика. 2024;(4):82-96. https://doi.org/10.37489/2588-0519-2024-4-82-96. EDN: MQOHEI
Оmelyanovskiy V.V., Kutsev S.I., Mukhortova P.A., Kharitonova A.G., Slabikova A.A., Ignatyeva N.V., Kingshott A.A., Teptsova T.S., Bogdanova V.O. Approaches to reduce the uncertainty in the clinical efficacy of orphan drugs. Kachestvennaya Klinicheskaya Praktika = Good Clinical Practice. 2024;(4):82-96. (In Russ.) https://doi.org/10.37489/2588-0519-2024-4-82-96. EDN: MQOHEI
НАШИ КНИГИ










Другие журналы
"Издательства ОКИ"
![]()
![]()
![]()
![]()
![]()
ПАРТНЕРЫ
![]()
Адрес редакции и издательства:
ООО «Издательство ОКИ»
121357, г. Москва, ул. Ватутина, д. 4, к. 2, кв. 22
ООО «Издательство ОКИ»
Генеральный директор Афанасьева Елена Владимировна
Тел. + 7 (916) 986-04-65; Email: eva88@list.ru
Обработка персональных данных
































